係丏栻棟妛揑抦幆
侾
僆僺僆僀僪
侾丏僆僺僆僀僪偲偼壗偐-栻棟妛揑摿挜
❶ 僆僺僆僀僪偲偼
僆僺僆僀僪乮opioid乯偲偼丆杻栻惈捔捝栻傗偦偺娭楢崌惉捔捝栻側偳偺傾儖僇儘僀僪偍傛傃儌儖僸僱條妶惈傪桳偡傞撪場惈傑偨偼崌惉儁僾僠僪椶偺憤徧偱偁傞丅
婭尦慜傛傝働僔枹弉壥偐傜嵦庢偝傟偨傾僿儞乮opium乯偑捔捝栻偲偟偰梡偄傜傟丆19 悽婭弶摢偵偼丆偦偺庡惉暘偲偟偰儌儖僸僱偑弶偺傾儖僇儘僀僪偲偟偰扨棧偝傟偨丅1970 擭戙偵偼丆僆僺僆僀僪偺嶌梡揰偲偟偰庴梕懱偑懚嵼偡傞偙偲偑徹柧偝傟丆弶傔偰栻暔庴梕懱偺奣擮偲偟偰摫擖偝傟偨丅偦偺屻丆撪場惈儌儖僸僱條暔幙偺扵嶕偑峴傢傟丆僄儞働僼傽儕儞丆僄儞僪儖僼傿儞丆僟僀僲儖僼傿儞丆嵟嬤偱偼僄儞僪儌儖僼傿儞側偳偑扨棧丒摨掕偝傟偨丅1990 擭戙偵偼丆兪丆兟偍傛傃內僆僺僆僀僪庴梕懱偺堚揱巕偑扨棧惛惢乮僋儘乕僯儞僌乯偝傟丆偦偺峔憿傗婡擻偑暘巕儗儀儖偐傜柧傜偐偵偝傟偰偄傞丅
❷ 僆僺僆僀僪庴梕懱偺峔憿偲忣曬揱払乮恾1乯
兪丆兟偍傛傃內僆僺僆僀僪庴梕懱偼丆偡傋偰GTP 寢崌抈敀幙乮G 抈敀幙乯仏1 偲嫟栶偡傞7 夞枌娧捠宆庴梕懱乮GPCR乯偱偁傞丅偙傟傜僆僺僆僀僪庴梕懱僞僀僾娫偺憡摨惈偼崅偔乮慡懱偱栺60亾乯丆摿偵嵶朎枌娧捠椞堟偱偼旕忢偵崅偄丅偄偢傟偺庴梕懱傕婎杮揑偵Gi/o 抈敀幙仏2 偲娭楢偟偰偍傝丆僆僺僆僀僪庴梕懱妶惈壔偵傛傝丆偝傑偞傑側嵶朎撪忣曬揱払宯偑塭嬁傪庴偗傞偙偲偵傛傝丆恄宱揱払暔幙偺梀棧傗恄宱嵶朎懱偺嫽暠惈偑掅壓偡傞偨傔偵恄宱嵶朎偺妶摦偑梷惂偝傟傞乮恾1乯丅
堦曽丆嬤擭丆儌儖僸僱偵傛傞捔捝岠壥敪尰偵偍偗傞嫽暠惈恄宱揱払偺娭梌傕帵偝傟丆壓峴惈梷惂宯仏3 偺捈愙揑妶惈壔傗丆嵶朎撪忣曬揱払宯傪妶惈壔偡傞偙偲偱捔捝岠壥傪敪尰偟偰偄傞偙偲傕柧傜偐偵偝傟偰偄傞乮恾1乯丅
儌儖僸僱丆僆僉僔僐僪儞丆僼僃儞僞僯儖丆僩儔儅僪乕儖乮惓妋偵偼戙幱嶻暔偺儌僲-O-扙儊僠儖懱丆埲壓M1乯偼丆偡傋偰兪僆僺僆僀僪庴梕懱偵懳偡傞恊榓惈偑崅偄傕偺偺丆偦傟偧傟偺栻暔娫偵偍偄偰丆擣傔傜傟傞栻棟嶌梡偵堘偄偑偁傞偙偲偑抦傜傟偰偄傞乮Ⅱ-4-1-6 奺僆僺僆僀僪偺栻棟妛揑摿挜偺崁嶲徠乯丅偙傟傜偺栻暔娫偵偍偗傞栻棟嶌梡偺堘偄偵娭偟偰偼丆偝傑偞傑側尒夝偑側偝傟偰偍傝丆兪僆僺僆僀僪庴梕懱偼兪1 偍傛傃兪2 庴梕懱丆兟僆僺僆僀僪庴梕懱偼兟1 偍傛傃兟2 庴梕懱丆內僆僺僆僀僪庴梕懱偼內1丆內2丆內3 庴梕懱側偳偺僒僽僞僀僾偺懚嵼偑採彞偝傟偰偒偨丅偟偐偟丆兪丆兟偍傛傃內僆僺僆僀僪庴梕懱傪僐乕僪偡傞堚揱巕偼偦傟偧傟1 庬椶偟偐懚嵼偟側偄偨傔丆僗僾儔僀僗僶儕傾儞僩仏4 埶懚惈僒僽僞僀僾傗僆僺僆僀僪庴梕懱偺懡検懱壔仏5 偵懳偡傞廋忺偺嵎堎丆偁傞偄偼僆僺僆僀僪庴梕懱偵懳偡傞棫懱峔憿曄宍偵婎偯偄偨儕僈儞僪仏6 埶懚惈僒僽僞僀僾側偳偺怴偟偄壖愢乮ligand biased efficacy 壖愢仏7乯偑採彞偝傟偰偄傞丅
恾1丂僆僺僆僀僪儕僈儞僪偲僆僺僆僀僪庴梕懱傪夘偟偨嵶朎撪忣曬揱払宯
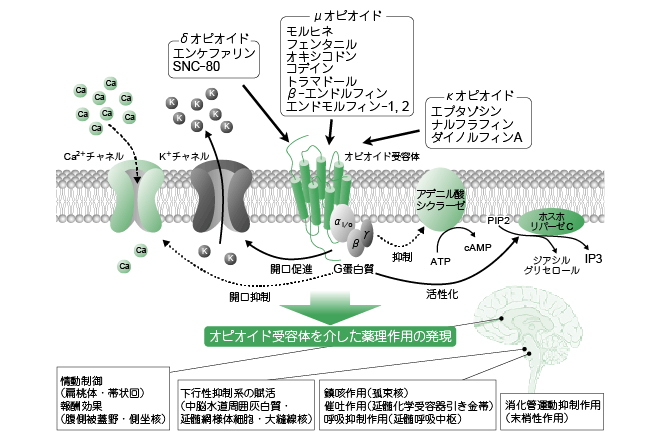
僆僺僆僀僪庴梕懱偺嵶朎撪忣曬揱払宯偼兪丆兟偍傛傃內僆僺僆僀僪庴梕懱偺娫偵戝偒側嵎偼懚嵼偟側偄偨傔丆偡傋偰偺僆僺僆僀僪庴梕懱偺嵶朎撪忣曬揱払傪傑偲傔偰婰嵹偟偨丅側偍丆僆僺僆僀僪庴梕懱偼偡傋偰7 夞枌娧捠宆偺G 抈敀幙嫟栶宆庴梕懱偱偁傝丆偦偺嵶朎撪忣曬揱払宯偼G 抈敀幙傪夘偟偰恑峴偡傞丅恾拞偺PIP2 偼儂僗僼傽僠僕儖僀僲僔僩乕儖擇儕儞巁傪丆IP3 偼僀僲僔僩乕儖嶰儕儞巁傪帵偡丅傑偨丆奺擼晹埵偱偺僆僺僆僀僪偵傛傞栻棟嶌梡傪恾壓偵傑偲傔偨丅
仏丗1丗GTP 寢崌抈敀幙乮G 抈敀幙乯
GTP 傾乕僛偵懏偡傞僌傾僯儞僰僋儗僆僠僪寢崌抈敀幙偺棯徧丅枌庴梕懱娭楢僿僥儘嶰検懱G 抈敀幙偲掅暘巕検G抈敀幙偑偁傞偑丆偙偙偱偼慜幰偺嶰検懱G 抈敀幙傪巜偡丅嶰検懱G 抈敀幙偼兛丆兝偍傛傃兞僒僽儐僯僢僩偐傜側傝丆G 抈敀幙嫟栶宆庴梕懱偑巋寖偝傟傞偲兛僒僽儐僯僢僩偵寢崌偟偰偄傞GDP 偲GTP 偺岎姺斀墳偑婲偙傝丆GTP 寢崌宆兛僒僽儐僯僢僩偲兝兞僒僽儐僯僢僩偵夝棧偡傞丅偙傟傜偺僒僽儐僯僢僩偼丆偦傟傜偺昗揑抈敀幙丒峺慺傪妶惈壔偟丆僔僌僫儖傪壓棳傊偲揱払偡傞丅
仏2丗Gi/o 抈敀幙
嶰検懱G 抈敀幙偼兛僒僽儐僯僢僩偺婡擻偍傛傃堚揱巕偺憡堘偐傜丆Gs丆Gi丆Go丆Gq丆Gt丆Golf 側偳偺僒僽僼傽儈儕乕偵暘椶偝傟偰偄傞丅Gi 偼傾僨僯儖巁僔僋儔乕僛傪梷惂偟丆Go 偼恄宱慻怐偵懡偔敪尰偟偰偄傞丅傑偨丆Gi/o 抈敀幙偐傜夝棧偟偨兝兞 僒僽儐僯僢僩偼丆K亄僠儍僱儖偺奐岥懀恑丆Ca2亄僠儍僱儖偺奐岥梷惂偲偄偭偨嵶朎撪墳摎傪堷偒婲偙偡丅
仏3丗壓峴惈梷惂宯
擼偐傜愐悜傪壓峴偟丆捝妎忣曬偺揱払傪梷惂偡傞宯丅擼偐傜愐悜傊恄宱揱払暔幙偺僲儖傾僪儗僫儕儞偲僙儘僩僯儞偑曻弌偝傟偰梷惂偡傞丅
仏4丗僗僾儔僀僗僶儕傾儞僩
RNA 慜嬱懱拞偺僀儞僩儘儞傪彍嫀偟丆慜屻偺僄僋僜儞傪嵞寢崌偡傞峴掱偱惗偠傞懡條側mRNA 偵傛傝惗惉偝傟傞抈敀幙孮丅
仏5丗庴梕懱偺懡検懱壔
嵶朎枌偵懚嵼偡傞庴梕懱偼丆1 暘巕偵傛偭偰傕丆嵶朎奜偺巋寖傪庴梕偟丆偦偺忣曬傪嵶朎撪傊揱払偡傞偙偲偑壜擻偱偁傞偑丆暋悢偺暘巕偑夛崌乮懡検懱壔乯偡傞偙偲偱丆堎側偭偨嵶朎撪忣曬揱払暘巕偑妶惈壔偝傟傞偨傔丆懡嵤側忣曬揱払偑壜擻偵側偭偰偄傞丅
仏6丗儕僈儞僪
庴梕懱傗峺慺偵寢崌偟丆惗暔妶惈傪堷偒婲偙偡暔幙丅峺慺偵懳偡傞婎幙丆曗峺慺丆栻暔乮庴梕懱嶌摦栻傗幷抐栻乯丆儂儖儌儞丆僒僀僩僇僀儞丆恄宱揱払暔幙側偳丅
仏7丗ligand biased efficacy 壖愢
栻暔乮儕僈儞僪乯偺寢崌偡傞庴梕懱偑摨堦偱偁偭偰傕丆儕僈儞僪偲庴梕懱偑宍惉偡傞暋崌懱偺棫懱峔憿偑堎側傞偨傔偵丆妶惈壔偝傟傞嵶朎撪墳摎偑儕僈儞僪偵埶懚偟偰堎側傞偲偄偆壖愢丅
❸ 僆僺僆僀僪庴梕懱傪夘偟偨栻棟嶌梡乮恾1丆昞1乯
儌儖僸僱丆僆僉僔僐僪儞丆僼僃儞僞僯儖丆僩儔儅僪乕儖側偳懡偔偺僆僺僆僀僪偵傛傞捔捝嶌梡偼丆庡偵兪僆僺僆僀僪庴梕懱傪夘偟偰敪尰偡傞丅兪僆僺僆僀僪庴梕懱傪夘偟偨捔捝嶌梡偼丆愐悜偵偍偗傞姶妎恄宱偵傛傞捝妎揱払偺梷惂傗帇彴傗戝擼旂幙抦妎椞堟側偳偺擼撪捝妎忣曬揱摫宱楬偺嫽暠梷惂偲偄偭偨忋峴惈捝妎忣曬揱払偺梷惂偵壛偊丆拞擼悈摴廃埻奃敀幙丆墑悜栐條懱嵶朎偍傛傃戝朌慄妀偵嶌梡偟丆墑悜-愐悜壓峴惈僲儖傾僪儗僫儕儞偍傛傃僙儘僩僯儞恄宱偐傜側傞壓峴惈梷惂宯偺晩妶壔側偳偵傛傞丅傑偨丆僆僺僆僀僪庴梕懱偼滸搷懱傗懷忬夞丆暊懁旐奧栰丆懁嵖妀側偳偺晹埵偵崅枾搙偵懚嵼偟偰偄傞偙偲偐傜丆忣摦惂屼偵傕怺偔娭傢偭偰偄傞丅偝傜偵丆偦偺懠偺拞悤恄宱宯嶌梡偲偟偰屇媧梷惂嶌梡乮墑悜屇媧拞悤偺捈愙梷惂嶌梡乯丆捔奝嶌梡乮屒懇妀奝拞悤傊偺抦妎擖椡梷惂乯丆嵜揻嶌梡乲墑悜壔妛庴梕婍堷偒嬥懷乮chemoreceptor trigger zone丟CTZ乯傊偺捈愙嶌梡乴側偳偑丆枛徑恄宱宯傊偺嶌梡偲偟偰徚壔娗塣摦梷惂嶌梡乮挵娗枌恄宱憄偱傾僙僠儖僐儕儞梀棧梷惂乯側偳偑抦傜傟偰偄傞丅
兟偍傛傃內僆僺僆僀僪庴梕懱偺妶惈壔偵傛偭偰傕丆兪僆僺僆僀僪庴梕懱偺妶惈壔偲摨條偵捔捝嶌梡偑擣傔傜傟傞丅偟偐偟丆兪僆僺僆僀僪庴梕懱偺妶惈壔偼懡岾姶乮曬廣岠壥仏1乯偑惗偠傞偺偵懳偟偰丆內僆僺僆僀僪庴梕懱偱偼寵埆姶傪堷偒婲偙偟乮拞擼曈墢僪僷儈儞恄宱慜廔枛梷惂偵傛傞僪僷儈儞梀棧梷惂乯丆儌儖僸僱側偳偵傛傞惛恄埶懚仏2傪梷惂偡傞丅傑偨丆兟偍傛傃內僆僺僆僀僪庴梕懱偺妶惈壔偵傛傞屇媧梷惂嶌梡偼丆兪僆僺僆僀僪庴梕懱偵傛傞傕偺偲斾傋庛偄丅
| 庴梕懱僞僀僾 | 兪僆僺僆僀僪庴梕懱 | 兟僆僺僆僀僪庴梕懱 | 內僆僺僆僀僪庴梕懱 |
|---|---|---|---|
栻棟嶌梡 捔捝嶌梡 |
|
|
|
| 嵶朎撪忣曬揱払 | cAMP 嶻惗↓丒Ca2亄 僠儍僱儖↓丒K亄僠儍 僱儖↑乮Gi/oα埶懚揑乯 PLC 妶惈壔丒PKC 妶 惈壔乮Gβγ埶懚揑乯 |
cAMP 嶻惗↓丒Ca2亄 僠儍僱儖↓丒K亄僠儍 僱儖↑乮Gi/oα埶懚揑乯 PLC 妶惈壔丒PKC 妶 惈壔乮Gβγ埶懚揑乯 |
cAMP 嶻惗↓丒Ca2亄 僠儍僱儖↓丒K亄僠儍 僱儖↑乮Gi/oα埶懚揑乯 |
| 庡側敪尰晹埵 | 戝擼旂幙丆慄忦懱丆帇 彴丆帇彴壓晹丆拞擼丆 嫶—墑悜乮惵斄妀丆屒懇 妀乯丆愐悜丆堦師姶妎恄 宱側偳 |
戝擼旂幙丆慄忦懱丆懁 嵖妀丆拞擼側偳 |
慄忦懱丆懁嵖妀丆帇彴丆 帇彴壓晹丆拞擼丆嫶—墑 悜乮惵斄妀丆屒懇妀乯丆 愐悜側偳 |
乮戝郪嫥峅丆拞愳婱擵丆惉揷丂擭乯
仏丗1丗曬廣岠壥
擼撪偺曬廣宯乮僪僷儈儞恄宱宯乯偑丆梸媮偑枮偨偝傟偨帪傗曬廣傪摼傞偙偲傪婜懸偟偰峴摦偟偰偄傞帪偵妶惈壔偟丆夣偺姶妎乮懡岾姶丆摡悓姶側偳乯傪梌偊傞岠壥丅
仏2丗惛恄埶懚
師偺偆偪偄偢傟偐1 偮傪娷傓峴摦偵傛偭偰摿挜偯偗傜傟傞堦師惈偺枬惈恄宱惗暔妛揑幘姵丅①帺屓惂屼偱偒偢偵栻暔傪巊梡偡傞丆②徢忬乮捝傒乯偑側偄偵傕偐偐傢傜偢嫮敆揑偵栻暔傪巊梡偡傞丆③桳奞側塭嬁偑偁傞偵傕偐偐傢傜偢帩懕偟偰巊梡偡傞丆④栻暔偵懳偡傞嫮搙偺梸媮偑偁傞丅嶲徠丅
俀丏崙撪偱棙梡壜擻側僆僺僆僀僪偲偦偺摿挜
❶ 惢嵻偺摿挜
2014 擭5 寧尰嵼丆擔杮崙撪偱偑傫醬捝偵懳偟偰棙梡壜擻側僆僺僆僀僪惢嵻偺堦棗傪昞2 偵帵偡丅
乮孖嶳弐擵丆梋媨偒偺傒乯
| 堦斒柤 | 彜昳柤 | 嵻宍丒婯奿 丒擹搙 |
搳梌宱楬 乮揔墳撪乯 |
搳梌 娫妘 |
曻弌 婡峔 |
惢嵻 偲偟偰偺 Tmax仏1 乮h乯 乮mean亇SD乯 |
惢嵻 偲偟偰偺 敿尭婜 乮h乯 乮mean亇SD乯 |
摿 挜 |
|---|---|---|---|---|---|---|---|---|
儌儖僸僱 |
僇僨傿傾儞® |
僇僾僙儖丗 20 mg丒30 mg丒 60 mg 僗僥傿僢僋棻丗 30 mg丒60 mg丒 120 mg |
宱岥 |
24 |
彊曻惈 |
7.3±0.8 |
9.2±0.9 |
pH 埶懚宆偺曻弌惂屼枌偱僐乕僥傿儞僌偝傟偨捈宎1.0乣1.7 mm 偺彊曻惈梓棻偑僇僾僙儖傑偨偼僗僥傿僢僋偵廩揢偝傟偰偄傞丅 |
僺乕僈乕僪® |
忶丗 20 mg丒30 mg丒 60 mg丒120 mg |
宱岥 |
24 |
彊曻惈 |
6.3±4.1 |
21.6±5.9 |
儌儖僸僱棸巁墫偵丆曻弌惂屼枌偲偟偰悈梟惈旝棻巕傪暘嶶偝偣偨悈晄梟惈崅暘巕偑僐乕僥傿儞僌偝傟偰偄傞丅徚壔娗撪偱悈梟惈旝棻巕偑懍傗偐偵梟夝偟偰懡悢偺嵶岴傪宍惉偟丆pH 旕埶懚揑偵彊曻惈傪帵偡丅崅帀朾怘怘屻偵搳梌偡傞偲Cmax仏2 偍傛傃AUC仏3 偑尭彮丆Tmax偑抶墑偡傞偑丆怘慜60 暘搳梌偱偁傟偽怘帠偺塭嬁偼柍帇偱偒傞偨傔丆怘娫搳梌偲偝傟偰偄傞丅 | |
MS僐儞僠儞® |
忶丗 10 mg丒30 mg丒 60 mg |
宱岥 |
12 |
彊曻惈 |
2.7±0.8 |
2.58± |
崅媺傾儖僐乕儖傪僐乕僥傿儞僌偟偨儌儖僸僱棻巕傪埑弅偟偨峔憿偱丆挵娗撪偺悈暘偵傛傝彊乆 偵梟夝偝傟傞丅 |
|
MS 僣儚僀僗儘儞® |
僇僾僙儖丗 10 mg丒30 mg丒 60 mg |
宱岥 |
12 |
彊曻惈 |
1.9±1.3 |
ND |
捈宎0.6乣1 mm 偺彊曻惈梓棻傪僇僾僙儖偵廩揢偟偨惢嵻偱丆挵娗撪偺悈暘偵傛傝彊乆偵惢嵻拞偺儌儖僸僱偑梟夝偡傞丅 | |
儌儖儁僗® |
嵶棻丗 2亾 乮10 mg/0.5 g/曪丆僶儔乯 6亾 乮30 mg/0.5 g/曪丆僶儔乯 |
宱岥 |
12 |
彊曻惈 |
2.4乣2.8 |
6.9乣8.7 |
儌儖僸僱傪娷傓棻巕偵彊曻惈旂枌傪僐乕僥傿儞僌偟丆偦偺忋偐傜娒枴椏傪僐乕僥傿儞僌偟偨峔憿偱丆捈宎栺0.5 mm 偺嵶棻偱偁傞丅宱娗搳梌壜丅 | |
儌儖僸僱 |
儌儖僸僱墫巁墫 |
枛丒忶丗 10 mg |
宱岥 |
4 帪娫枅 |
懍曻 |
0.5乣1.3 |
2.0乣3.0 |
掕婜搳梌傑偨偼儗僗僉儏乕栻偲偟偰巊梡偡傞丅 |
僆僾僜® |
撪暈塼丗 5 mg/2.5 mL/曪 10 mg/5 mL/曪 |
宱岥 |
4 帪娫枅 |
懍曻惈 |
0.5±0.2 |
2.9±1.1 |
儌儖僸僱宱岥搳梌奐巒帪偺梡検挷愡偍傛傃梡検挷愡屻偺醬捝帯椕偵巊梡偱偒丆傑偨丆僆僺僆僀僪彊曻惈惢嵻搳梌拞偺儗僗僉儏乕栻偲偟偰傕巊梡偡傞丅 | |
僷僔乕僼® |
僇僾僙儖丗 30 mg丒60 mg丒 120 mg |
宱岥 |
24 |
彊曻惈 |
懍曻晹丗 |
11.3乣 |
懍曻惈嵶棻偲彊曻惈嵶棻偑僇僾僙儖偵廩揢偝傟丆1 擔1 夞搳梌偱搳梌屻憗婜偐傜24 帪娫埨掕偟偨捔捝岠壥傪堐帩偱偒傞傛偆偵愝寁偝傟偨惢嵻偱偁傞丅 | |
傾儞儁僢僋® |
嵖嵻丗 10 mg丒20 mg丒 30 mg |
捈挵撪 |
6乣12 帪娫枅 |
— |
1.3乣1.5 |
4.2乣6.0 |
媧廂偑懍傗偐偱丆搳梌屻栺8 帪娫傑偱桳岠寣拞擹搙偑曐偨傟傞丅 | |
儌儖僸僱 |
拲丗 乮儌儖僸僱丆傾儞儁僢僋®乯 乮僾儗儁僲儞®乯 |
乮儌儖僸僱丆傾儞儁僢僋®乯旂壓惷柆撪峝枌奜僋儌枌壓 乮僾儗儁僲儞®乯旂壓惷柆撪 |
扨夞 |
— |
惷柆撪丗 |
惷柆撪丗 |
僾儗儁僲儞® 偼僾儗僼傿儖僪僔儕儞僕偱偁傝丆拲幩嵻挷惢傗搳梌偺娙曋惈丒埨慡惈傪岦忋偝偣偨惢嵻偱偁傞丅桝塼嵻偵攝崌偟偰搳梌偡傞偐丆僔儕儞僕億儞僾傑偨偼実懷宆僨傿僗億乕僓僽儖拲擖億儞僾傪梡偄偰搳梌偡傞丅 | |
僆僉僔僐僪儞 |
僆僉僔僐儞僠儞® |
忶丗 5 mg丒10 mg丒 20 mg丒40 mg |
宱岥 |
12 |
彊曻惈 |
4.0±2.5 |
9.2±2.6 |
傾僋儕儖巁宯崅暘巕枌偲崅媺傾儖僐乕儖枌偺擇廳峔憿偱丆挵娗撪偺悈暘偑怹摟偟丆僆僉僔僐僪儞偑彊乆偵彫挵撪傊曻弌偝傟傞丅儅僩儕僢僋僗婎嵻乮敳偗妅乯偑暢曋拞偵攔煏偝傟傞応崌偑偁傞偑丆惉暘偼偡偱偵媧廂偝傟偰偄傞偨傔丆椪彴忋栤戣偼側偄丅 |
僆僉僲乕儉® |
嶶乮0.5亾乯丗 2.5 mg/0.5 g/曪 5 mg/1 g/曪 10 mg/1 g/曪 20 mg/1 g/曪 |
宱岥 |
6 帪娫枅 |
懍曻惈 |
1.7乣1.9 |
4.5乣6.0 |
僆僉僔僐僪儞宱岥惢嵻傪梡偄傞嵺偺梡検挷愡傗丆撍弌捝傊偺儗僗僉儏乕栻偲偟偰巊梡偡傞丅 | |
僆僉僼傽僗僩® |
拲丗 10 mg/1 mL/A 50 mg/5 mL/A |
惷柆撪 |
扨夞 丒帩懕 |
— |
媫懍扨夞 |
帩懕惷拲 |
— |
|
僼僃儞僞 |
僨儏儘僥僢僾® MT |
揬晅嵻丗 2.1 mg乮12.5μg/h乯 4.2 mg乮25μg/h乯 8.4 mg乮50μg/h乯 12.6 mg乮75μg/h乯 16.8 mg乮100μg/h乯 |
宱旂 |
72 帪娫枅 |
彊曻惈 |
30乣36 |
21乣23 |
儅僩儕僢僋僗僞僀僾偺宱旂媧廂宆惢嵻偱偁傞丅懠偺僆僺僆僀僪捔捝栻偐傜愗傝懼偊偰巊梡偡傞丅娷検偑堎側傞5 惢嵻乮2.1 mg丆4.2 mg丆8.4 mg丆12.6 mg丆16.8 mg 偑偁傝丆扨埵柺愊偁偨傝偺曻弌懍搙偼偄偢傟傕摨堦偱偁傞丅 |
儚儞僨儏儘® |
揬晅嵻丗 0.84 mg乮0.3 mg/擔乯 1.7 mg乮0.6 mg/擔乯 3.4 mg乮1.2 mg/擔乯 5 mg乮1.8 mg/擔乯 6.7 mg乮2.4 mg/擔乯 |
宱旂 |
24 帪娫枅 |
彊曻惈 |
18乣26 |
20.0乣22.4 |
72 帪娫枅偺惢嵻偲栻暔摦懺偑戝偒偔曄傢傜側偄丅 | |
僼僃儞僩僗® |
揬晅嵻丗 1 mg丒2 mg丒4 mg丒6 mg丒8 mg |
宱旂 |
24 帪娫枅 |
彊曻惈 |
20.1±6.1 |
25.7乣31.3 |
— |
|
僀乕僼僃儞® |
岥峯擲枌媧廂嵻 乮僶僢僇儖忶乯丗 50μg丒100μg 200μg丒400μg 600μg丒800μg |
宱岥峯擲枌 |
1 夞偺撍弌捝偵懳偟偰30暘埲忋偁偗偰1 夞偺傒捛壛壜擻 |
懍曻惈 |
0.59乣0.67 |
3.37乣 |
忋戝塒帟偺帟宻偲杍偲偺娫偵嫴傒偙傓傛偆偵偍偄偰丆梟夝偝偣傞丅掕婜揑側嫮僆僺僆僀僪偺搳梌傪庴偗偰偄傞姵幰傪懳徾偲偡傞丅尨懃丆儌儖僸僱宱岥姺嶼30噐/擔埲忋偺搳梌傪庴偗偰偄傞姵幰傪懳徾偲偡傞丅偦傟枹枮偺姵幰偱偼怲廳偵揔墳傪専摙偡傞丅50 傑偨偼100μg 偐傜奐巒偡傞丅1 夞800μg 巊梡偟偰傕岠壥偑晄廫暘側偄応崌偼懠偺曽朄傊偺曄峏傪専摙偡傞丅 | |
傾僽僗僩儔儖® |
岥峯擲枌媧廂嵻 乮愩壓忶乯丗 100μg丒200μg丒 400μg |
宱岥峯擲枌 |
1 夞偺撍弌捝偵懳偟偰30暘埲忋偁偗偰1 夞偺傒捛壛壜擻 |
懍曻惈 |
0.5乣1.0 |
5.02乣13.5 |
愩壓偵梟偐偟偰岥峯擲枌傛傝媧廂偝偣傞丅掕婜揑側嫮僆僺僆僀僪偺搳梌傪庴偗偰偄傞姵幰傪懳徾偲偡傞丅尨懃丆儌儖僸僱宱岥姺嶼60噐/擔埲忋偺搳梌傪庴偗偰偄傞姵幰傪懳徾偲偡傞丅偦傟枹枮偺姵幰偱偼怲廳偵揔墳傪専摙偡傞丅掕婜搳梌検偵偐偐傢傜偢丆100μg 偐傜奐巒偡傞丅1 夞800μg 巊梡偟偰傕岠壥偑晄廫暘側応崌偼懠偺曽朄傊偺曄峏傪専摙偡傞丅 | |
僼僃儞僞僯儖® |
拲丗 0.1 mg/2 mL/A 0.25 mg/5 mL/A 0.5 mg/10 mL/A |
惷柆撪 峝枌奜 僋儌枌壓 |
惷丒峝丗 僋儌枌壓丗 |
— |
惷柆撪丗 峝枌奜丗 |
惷柆撪丗 |
— |
|
儁僠僕儞 |
僆僺僗僞儞® |
枛 | 宱岥 |
8 |
懍曻惈 |
2.0 |
3.5 |
— |
僆僺僗僞-儞® |
拲丗 35 mg/1 mL/A 50 mg/1 mL/A |
旂壓 |
3乣4 帪娫枅 |
— |
嬝擏撪丗 |
嬝擏撪丗 |
||
| 僐僨僀儞 | 僐僨僀儞儕儞巁墫 |
嶶丗 10 mg/g乮1亾乯 100 mg/g乮10亾乯 忶丗 5 mg丒20 mg |
宱岥 |
4乣6 帪娫枅 |
懍曻惈 |
0.8±0.2 |
2.2±0.2 |
僐僨僀儞偼懱撪偱儌儖僸僱偵戙幱偝傟傞偙偲偵傛傝捔捝岠壥傪敪婗偡傞偲峫偊傜傟偰偄傞丅 |
| 僕僸僪儘僐僨僀儞 | 僕僸僪儘僐僨僀儞儕儞巁墫 |
枛丒嶶丗 10 mg/g乮1亾乯 100 mg/g乮10亾乯 |
宱岥 |
4乣6 帪娫枅 |
懍曻惈 |
1.6乣1.8 |
3.3乣3.7 |
僐僨僀儞偵斾傋偰捔捝嶌梡偼傎傏摨摍丅 |
僩儔儅僪乕儖 |
僩儔儅乕儖® |
僇僾僙儖丗 25 mg丒50 mg |
宱岥 |
4乣6 帪娫枅 |
懍曻惈 |
僩儔儅僪乕儖丗 |
僩儔儅僪乕儖丗 |
娞忈奞丒恡忈奞姵幰偱偼Cmax丆AUC0乣∞丆T1/2偑墑挿偡傞丅 |
僩儔儅乕儖® |
拲丗 100 mg/2 mL |
嬝擏撪 |
4乣5 帪娫枅 |
— |
ND |
ND |
僆僺僆僀僪嶌梡偍傛傃儌僲傾儈儞憹嫮嶌梡偵傛傝捔捝岠壥傪帵偡丅CYP2D6 偵傛偭偰戙幱偝傟傞M1 偑μ僆僺僆僀僪庴梕懱偺恊榓惈偑崅偄丅儌僲傾儈儞嵞庢傝崬傒慾奞嶌梡偼M1 傛傝僩儔儅僪乕儖偺傎偆偑崅偄丅 | |
僽僾儗僲儖僼傿儞 |
儗儁僞儞® |
嵖嵻丗 0.2 mg丒0.4 mg |
捈挵撪 |
8乣12 帪娫枅 |
— |
1.0乣2.0 |
ND |
杻栻漢峈惈捔捝栻仏4 |
| 拲丗 0.2 mg/1 mL/A 0.3 mg/1.5 mL/A |
嬝擏撪 |
6乣8 帪娫枅 |
— |
亙0.08 |
2乣3 |
|||
| 儁儞僞僝 僔儞 |
僜僙僑儞® |
忶丗 25 mg |
宱岥 |
3乣5 帪娫枅 |
懍曻惈 |
2.0 |
1.6乣3.2 |
杻栻漢峈惈捔捝栻 忶嵻偵偼丆晄揔愗側巊梡朄傪杊巭偡傞偨傔偵杻栻漢峈栻偱偁傞僫儘僉僜儞墫巁墫偑揧壛偝傟偰偄傞丅 |
| 拲丗 15 mg/1 mL/A 30 mg/1 mL/A |
旂壓 |
3乣4 帪娫枅 |
— |
嬝拲丗 |
嬝拲丗 |
|||
| 僄僾僞僝僔儞 | 僙僟儁僀儞® |
拲丗 15 mg/1 mL/A |
旂壓 |
扨夞 |
— |
旂壓丒嬝 |
旂壓丒嬝 |
杻栻漢峈惈捔捝栻 |
| 儊僒僪儞 | 儊僒儁僀儞® |
忶丗 5 mg丒10 mg |
宱岥 |
8 帪娫枅 |
懍曻惈 |
4.9±2.1 |
37.2±4.6 |
偁傞僆僺僆僀僪偵懳偡傞懴惈傪桳偟偰偄偰傕丆岎嵎懴惈偑晄姰慡側応崌偑偁傞丅姺嶼斾傕堦掕偟偨傕偺偑側偔丆夁検搳梌偵廫暘側拲堄偑昁梫偱偁傞丅 |
| 僞儁儞僞 僪乕儖 |
僞儁儞僞® |
忶丗 25 mg丒50 mg丒 100 mg |
宱岥 |
12 帪娫枅 |
彊曻惈 |
5 |
5乣6 |
晄惓巊梡杊巭傪栚揑偵億儕僄僠儗儞僆僉僒僀僪偑巊梡偝傟偨忶嵻乮TRF乯偱丆僴儞儅乕傪巊梡偟偰傕夡傟側偄峔憿偵側偭偰偄傞丅 |
- 仏1丗
- Tmax乮maximum drug concentration time乯丟嵟崅寣拞擹搙摓払帪娫丅栻暔搳梌屻丆寣拞擹搙偑嵟戝乲嵟崅寣拞擹搙乮Cmax乯乴偵摓払偡傞傑偱偺帪娫丅
- 仏2丗
- Cmax乮maximum drug concentration乯丟嵟崅乮嵟戝乯寣拞擹搙丅栻暔搳梌屻偺寣拞擹搙偺嵟戝抣丅
- 仏3丗
- AUC乮area under the drug concentration time curve乯丟栻暔寣拞擹搙乮帪娫乯嬋慄壓柺愊丅栻暔寣拞擹搙傪宱帪揑偵昞偟偨嬋慄僌儔僼偲帪娫幉乮墶幉乯偵埻傑傟偨晹暘偺柺愊丅寣拞偵庢傝崬傑傟偨栻偺検乮媧廂棪乯偺巜昗偲偟偰梡偄傞丅
- 仏4丗
- 杻栻漢峈惈捔捝栻丟僆僺僆僀僪嶌摦栻偑懚嵼偟側偄忬嫷偱偼嶌摦栻偲偟偰嶌梡偡傞偑丆僆僺僆僀僪嶌摦栻偺懚嵼壓偱偼偦偺嶌梡偵漢峈偡傞嶌梡傪傕偮捔捝栻丅
俁丏搳梌宱楬偺曄峏
僆僺僆僀僪偺婎杮揑側搳梌宱楬偼宱岥偩偑丆岥撪墛丆殝壓崲擄丆徚壔娗暵嵡丆埆怱丒歲揻側偳偺尨場偐傜宱岥搳梌偑宲懕偱偒偢丆搳梌宱楬偺曄峏偑昁梫偲側傞応崌偑偁傞丅戙懼宱楬偲偟偰偼捈挵撪搳梌丆宱旂搳梌丆旂壓丒惷柆撪搳梌偑偁傞丅拲幩偺応崌偵偼堦斒揑偵帩懕搳梌偑峴傢傟傞丅偦傟偧傟巊梡偱偒傞栻暔偺庬椶丆嵻宍偵尷傝偑偁傝丆傑偨搳梌宱楬偵傛傞摿挜傕堎側傞偺偱屄乆偺姵幰偵偁傢偣偰慖戰偡傞丅
❶ 宱岥搳梌
怤廝偑側偔丆娙曋偱宱嵪揑偱偁傝丆僆僺僆僀僪搳梌偱偼婎杮偺搳梌宱楬偲偝傟傞丅撪暈偟偨栻嵻偼挵娗偐傜媧廂偝傟傞嵺丆挵娗偺峺慺偵傛偭偰偁傞掱搙戙幱偝傟丆偝傜偵娞憻偱偺弶夞捠夁岠壥乮娞弶夞捠夁岠壥仏1乯傪庴偗傞丅偦偺偨傔偵懠偺宱楬偲斾妑偡傞偲搳梌検偼懡偔昁梫偱丆儌儖僸僱偱偼戙幱嶻暔乲儌儖僸僱-6-僌儖僋儘僯僪 乮M6G乯仏2丆儌儖僸僱-3-僌儖僋儘僯僪乮M3G乯仏3乴偑懡偔側傞丅
岥撪墛丆殝壓忈奞丆徚壔娗暵嵡丆埆怱丒歲揻丆偣傫栂側偳偱搳梌宲懕偑崲擄側応崌偼懠偺搳梌宱楬偵曄峏偡傞丅
❷ 捈挵撪搳梌
搳梌偼斾妑揑娙曋偱丆媧廂傕懍傗偐偱偁傞偑丆搳梌偵晄夣姶傪敽偆偨傔丆挿婜揑側巊梡偼揔偝側偄偙偲偑偁傞丅
捈挵墛丆壓棢丆汨栧丒捈挵偵憂晹偑懚嵼偡傞応崌丆廳搙偺寣彫斅尭彮丒敀寣媴尭彮帪偼搳梌傪旔偗傞丅
恖岺汨栧傪憿愝偟偰偄傞姵幰偺応崌丆恖岺汨栧偐傜偺搳梌偼丆偦偺惗懱撪棙梡棪偵偽傜偮偒偑偁傞偲曬崘偝傟偰偍傝丆挿婜揑側巊梡偼悇彠偝傟側偄丅惷柆憄偑朢偟偄偨傔媧廂偑埆偔晄埨掕偱丆栻嵻偑曋偲崿偠傝傗偡偔丆攔弌偺挷愡傕崲擄側偙偲側偳偑棟桼偲峫偊傜傟偰偄傞丅
❸ 宱旂搳梌
24 帪娫丒72 帪娫嶌梡偑帩懕偡傞僼僃儞僞僯儖揬晅嵻偑巊梡偝傟偰偄傞丅偙偺惢嵻偱偺岠壥偺敪尰偼揬晅奐巒屻12乣14 帪娫屻偱偁傝丆揬晅拞巭屻乮剝棧屻乯16乣24 帪娫偼捔捝岠壥偑帩懕偡傞偺偱丆搳梌奐巒帪娫傗拞巭帪娫偵拲堄偡傞丅
恦懍側搳梌検偺曄峏偑擄偟偄偨傔丆尨懃偲偟偰醬捝僐儞僩儘乕儖偺埨掕偟偰偄傞応崌偵巊梡偡傞丅撍弌捝偵懳偟偰偼懠偺搳梌宱楬偱偺僆僺僆僀僪搳梌偑昁梫偲側傞丅
揬晅晹埵偺旂晢偺忬懺偑埆偄応崌丆敪娋偑懡偄応崌偼丆媧廂偑埨掕偟側偄偨傔搳梌傪旔偗傞丅傑偨丆揬晅晹埵偺壏搙忋徃偱僼僃儞僞僯儖偺曻弌偑憹偡偨傔丆敪擬偟偰偄傞姵幰傗揬晅晹埵偺壛壏偵拲堄偡傞丅
仏丗1丗娞弶夞捠夁岠壥
宱岥搳梌偟偨栻暔偼彫挵偱媧廂偝傟丆娞憻傪宱偰慡恎傪弞娐偡傞偑丆偙偺帪丆娞憻偵懚嵼偡傞懡偔偺峺慺偵傛偭偰栻暔偑戙幱偝傟傞偙偲丅宱岥嵻偼娞弶夞捠夁岠壥偑戝偒偄丅
仏2丗儌儖僸僱-6-僌儖僋儘僯僪乮M6G乯
儌儖僸僱偺戙幱嶻暔偺堦偮丅嫮椡側捔捝嶌梡傪桳偡傞丅擼堏峴惈偑儌儖僸僱傛傝傕掅偔丆備偭偔傝偲寣塼擼娭栧傪捠夁偡傞偨傔偵嶌梡帩懕帪娫偑挿偄丅
仏3丗儌儖僸僱-3-僌儖僋儘僯僪乮M3G乯
儌儖僸僱偑娞憻偱戙幱偝傟偰惗偠傞嶻暔偺堦偮丅捔捝妶惈偼側偄偑丆恄宱撆惈傪桳偟偰偄傞偲偺曬崘傕偁傞丅
仏4丗儗僗僉儏乕栻
醬捝帪偵椪帪偵捛壛偡傞椪帪捛壛搳梌栻丅
❹ 帩懕旂壓拲
帩懕惷拲偲斾傋偰怤廝偑彮側偔丆埨慡偱娙曋側搳梌宱楬偱偁傞丅搳梌検偺曄峏偑恦懍偵峴偊傞偺偱丆醬捝僐儞僩儘乕儖偺晄埨掕側応崌傗媫懍側梡検偺挷惍傪昁梫偲偡傞応崌偵椙偄揔墳偲側傞丅旂壓傊偺搳梌懍搙偺忋尷偼堦斒揑偵1 mL/h 偲偝傟偰偄傞丅儗僗僉儏乕栻仏4 偲偟偰憗憲傝偟偨応崌偵傕丆捝傒傪惗偠側偄棳検偱偺巊梡傪峫椂偟丆旂壓慻怐偵巋寖乮捝傒傗夡巰側偳乯偑偁傞栻嵻偼旔偗傞丅
❺ 帩懕惷拲
丂妋幚丒恦懍側岠壥乮嵟戝岠壥偼5乣15 暘乯偑摼傜傟傞丅懠偺宱楬偱偼崲擄側戝検偺僆僺僆僀僪搳梌傕壜擻偱偁傞丅
帩懕旂壓拲偑偱偒側偄応崌乮恓偺巋擖晹偵擽釃丆敪愒丆峝寢偑偱偒傞乯丆嬅屌擻偺忈奞偑偁傞応崌丆偡偱偵惷柆儔僀儞偑偁傞応崌偵揔墳偲側傞丅丅
❻ 嬝擏撪搳梌
媧廂偑晄埨掕偱丆搳梌偺嵺偵捝傒偑嫮偄偨傔巊梡偟側偄丅旂壓搳梌丆帩懕旂壓拲丒帩懕惷拲傪梡偄傞丅
❼ 宱岥峯擲枌搳梌
僼僃儞僞僯儖岥峯擲枌媧廂嵻偑巊梡偝傟偰偄傞丅杮嵻偼撍弌捝偵懳偡傞儗僗僉儏乕栻偲偟偰梡偄傜傟傞丅宱岥搳梌偵斾傋偰媧廂偑懍傗偐側偺偑摿挜偱偁傞丅僼僃儞僞僯儖偼宱岥搳梌傪峴偆偲惗懱撪棙梡棪仏1 偑掅壓偡傞丅偙偺偨傔姎傑偢偵岥峯擲枌偐傜媧廂偝偣傞昁梫偑偁傞丅
仏丗1丗惗懱撪棙梡棪
搳梌偟偨栻暔偺壗亾偑惗懱撪乮寣拞乯偵庢傝崬傑傟丆柍懯側偔妶梡偝傟傞偐偲偄偆栻暔偺棙梡棪乮媧廂棪乯丅惗暔妛揑棙梡棪丆僶僀僆傾儀僀儔價儕僥傿乮bioavailability乯偲傕偄偆丅
係丏僆僺僆僀僪僗僀僢僠儞僌
❶ 僆僺僆僀僪僗僀僢僠儞僌
乵掕丂媊乶丂僆僺僆僀僪僗僀僢僠儞僌偲偼丆僆僺僆僀僪偺暃嶌梡偵傛傝捔捝岠壥傪摼傞偩偗偺僆僺僆僀僪傪搳梌偱偒側偄帪傗丆捔捝岠壥偑晄廫暘側帪偵丆搳梌拞偺僆僺僆僀僪偐傜懠偺僆僺僆僀僪偵曄峏偡傞偙偲傪偄偆丅
僆僺僆僀僪偺搳梌宱楬偺曄峏傪僆僺僆僀僪僗僀僢僠儞僌偵娷傓応崌偑偁傞偑丆杮僈僀僪儔僀儞偱偼栻暔偺曄峏偺傒傪僆僺僆僀僪僗僀僢僠儞僌偲掕媊偡傞丅
乵揔丂墳乶丂僆僺僆僀僪僗僀僢僠儞僌傪峴偆揔墳偼丆壓婰偺偲偍傝偱偁傞丅
- ①暃嶌梡偑嫮偔僆僺僆僀僪偺搳梌偺宲懕傗憹検偑崲擄側応崌
- ②捔捝岠壥偑晄廫暘側応崌
乮1乯暃嶌梡偑嫮偔僆僺僆僀僪偺憹検丒宲懕偑崲擄側応崌
僆僺僆僀僪僗僀僢僠儞僌偵傛傝丆尰嵼搳梌拞偺僆僺僆僀僪傗偦偺戙幱暔偵傛傝堷偒婲偙偝傟偰偄傞暃嶌梡乮偣傫栂丆柊婥丆尪妎丆埆怱丒歲揻丆曋旈側偳乯偑夵慞偡傞偙偲偑抦傜傟偰偄傞丅崅搙側恡婡擻忈奞偺偁傞姵幰偱丆儌儖僸僱傪巊梡偟偨応崌丆戙幱嶻暔偱偁傞M6G丆M3G 偺攔煏偑掅壓偟偰拁愊偟暃嶌梡偑弌尰偟傗偡偄壜擻惈偑偁傝丆僆僉僔僐僪儞丆僼僃儞僞僯儖傊偺曄峏偑桳岠側応崌偑偁傞丅
乮2乯捔捝岠壥偑晄廫暘側応崌
摨偠僆僺僆僀僪傪搳梌偟懕偗偨応崌丆懴惈偑惗偠偰丆堦掕検偺僆僺僆僀僪偵傛偭偰摼傜傟傞捔捝岠壥偑尭庛偟丆僆僺僆僀僪傪憹検偟偰傕捔捝岠壥偑摼傜傟側偄偙偲偑偁傞丅僆僺僆僀僪僗僀僢僠儞僌傪峴偆偲捔捝岠壥偑揔愗偵敪婗偝傟丆醬捝帯椕偵昁梫側僆僺僆僀僪偺搳梌検傕尭傜偡偙偲偑偱偒傞応崌偑偁傞丅偙傟偼丆堎側傞僆僺僆僀僪娫偱偼岎嵎懴惈偑晄姰慡仏2 側偨傔偲峫偊傜傟偰偄傞丅
仏2丗晄姰慡側岎嵎懴惈
僆僺僆僀僪娫偱偼丆岎嵎懴惈偑晄姰慡偱偁傞丅
岎嵎懴惈偲偄偆偺偼偁傞惗暔偑丆1 庬椶偺栻暔偵懳偟偰懴惈傪妉摼偡傞偲摨帪偵丆摨偠傛偆側峔憿傪傕偮暿偺庬椶偺栻嵻偵懳偡傞懴惈傕妉摼偟偰偟傑偆偙偲傪偄偆丅堎側傞僆僺僆僀僪娫偱偼偙偺岎嵎懴惈偑晄姰慡偱偁傞偨傔丆巊梡偟偰偄偨1 庬椶偺僆僺僆僀僪偵懳偟偰偁傞姵幰偑懴惈傪妉摼偟丆捔捝岠壥偑掅壓偟偨応崌偱傕丆僆僺僆僀僪偺庬椶傪曄峏偡傞偙偲偵傛偭偰丆捔捝岠壥偺夞暅傪婜懸偱偒傞偲峫偊傜傟傞丅
偦偺偨傔丆僆僺僆僀僪僗僀僢僠儞僌偱偼怴偨側僆僺僆僀僪偑丆寁嶼忋摍椡壙偲側傞姺嶼検傛傝傕彮検偱桳岠側偙偲偑偁傞丅堦曽丆夁検搳梌偲側偭偨傝丆偡偱偵懴惈偑偱偒偰偄偨柊婥側偳偺暃嶌梡偑嵞弌尰偡傞偙偲傕偁傞丅
❷ 僆僺僆僀僪僗僀僢僠儞僌偺幚嵺
婎杮揑側曽朄偼埲壓偵弎傋傞偲偍傝偱偁傞丅僆僺僆僀僪僗僀僢僠儞僌偼姵幰偺忬懺偵傛偭偰嵶傗偐側挷惍偑昁梫偱偁傞偨傔丆廫暘側宱尡傪傕偨側偄応崌偼丆娚榓働傾僠乕儉側偳偺愱栧壠偵憡択偡傞偙偲偑朷傑偟偄丅
- ①姺嶼偡傞僆僺僆僀僪偺丆寁嶼忋摍椡壙偲側傞姺嶼検傪媮傔傞丅姺嶼昞乮昞3乯偵廬偄丆尰嵼偺僆僺僆僀僪偲怴偟偄僆僺僆僀僪偺1 擔搳梌検傪寁嶼偡傞丅尰嵼偺僆僺僆僀僪偺搳梌偑斾妑揑戝検偱偁傞応崌偼丆堦搙偵曄峏偣偢悢夞偵暘偗偰僆僺僆僀僪僗僀僢僠儞僌傪峴偆丅
- ②姵幰偺忬懺偵偁傢偣偰丆栚昗偲偡傞姺嶼検傪愝掕偡傞丅寁嶼忋偺姺嶼検偼乽栚埨乿偱偁傝丆僆僺僆僀僪娫偺晄姰慡側岎嵎懴惈傗丆栻暔偵懳偡傞斀墳偺屄懱嵎偑戝偒偄偙偲偐傜丆幚嵺偵偼姺嶼昞偳偍傝偵側傜側偄偙偲傪峫椂偟丆姵幰屄恖偵偁傢偣偨搳梌検傊挷惍偡傞偙偲偑廳梫偱偁傞丅堦斒揑偵丆醬捝僐儞僩儘乕儖偼椙岲偩偑丆暃嶌梡偺偨傔偵僆僺僆僀僪僗僀僢僠儞僌傪峴偆応崌偼丆慜弎偺晄姰慡側岎嵎懴惈偺懚嵼偵傛傝丆寁嶼忋摍椡壙偲側傞検傛傝傕彮側偄検偱捔捝偑堐帩偱偒傞応崌偑偁傞偺偱拲堄傪梫偡傞丅傑偨丆姵幰偺昦忬偑埆偄丆崅楊偱偁傞側偳偺応崌傕丆彮検偐傜偺曄峏偑朷傑偟偄丅
- ③捔捝岠壥偺敪尰帪娫丆嵟戝岠壥偺帪娫丆帩懕帪娫傪峫椂偟偰丆怴偟偄僆僺僆僀僪偺搳梌奐巒帪娫丆搳梌娫妘傪寛掕偡傞丅捝傒偺憹嫮偺壜擻惈傕峫椂偟偰丆儗僗僉儏乕栻偺巜帵傪峴偆丅
- ④僆僺僆僀僪僗僀僢僠儞僌屻偺姵幰偺捝傒傗暃嶌梡偺憹尭傪拲堄怺偔娤嶡偟丆嵟揔側搳梌検傪寛掕偡傞丅
乵拲丂堄乶
- 僆僉僔僐僪儞丆僼僃儞僞僯儖偐傜儌儖僸僱偵曄峏偡傞応崌丆恡婡擻忈奞偺偁傞姵幰偱偼暃嶌梡傪惗偠傞応崌偑偁傞偨傔丆彮検偐傜奐巒偟偰廫暘偵娤嶡偡傞丅
- 儌儖僸僱偐傜僼僃儞僞僯儖傊偺曄峏偱偼挵寰摦偺槾恑偑婲偙傞偙偲偑懡偄偨傔丆娚壓栻偺尭検側偳偑昁梫側偙偲偑偁傞丅
俆丏姺嶼昞
姺嶼斾偵娭偟偰偼懡偔偺曬崘偑側偝傟偰偍傝丆偦偺悢抣偵偼偽傜偮偒偑偁傞丅傑偨丆懡偔偺曬崘偼捝傒偺埨掕偟偰偄傞姵幰偱偺懳儌儖僸僱偱偺扨夞搳梌偺寢壥偵婎偯偄偨姺嶼偲側偭偰偄傞丅幚嵺偺恌椕偱偼丆捝傒偺晄埨掕側姵幰偱偺曄峏偑懡偔丆姺嶼昞偺傒偵棅偭偨曄峏偼偡傞傋偒偱偼側偄丅姺嶼昞傪栚埨偵寛掕偟偨曄峏屻偺搳梌検偐傜丆屄乆偺姵幰偺捝傒丆暃嶌梡傪娤嶡偟偨偆偊偱偒傔嵶偐偄挷愡傪偡傞偙偲偑昁梫偱偁傞丅
杮僈僀僪儔僀儞偱偼昗弨揑側姺嶼偺栚埨偲偟偰丆奺庬僈僀僪儔僀儞側偳偺姺嶼昞傪傕偲偵専摙偟丆巊梡偟傗偡偄偲巚傢傟傞悢抣傪帵偡偙偲偲偟偨乮昞3乯丅
| 搳梌宱楬 | 惷柆撪搳梌丒旂壓搳梌 | 宱岥搳梌 | 捈挵撪搳梌 | 宱旂搳梌 |
|---|---|---|---|---|
儌儖僸僱 |
10乣15 mg | 30 mg | 20 mg | |
僐僨僀儞 |
200 mg | |||
僩儔儅僪乕儖 |
150 mg | |||
僆僉僔僐僪儞 |
15 mg | 20 mg | ||
僼僃儞僞僯儖 |
0.2乣0.3 mg | 仸 |
儌儖僸僱宱岥30 mg 傪婎弨偲偟偨応崌偵丆寁嶼忋摍椡壙偲側傞僆僺僆僀僪偺姺嶼検傪帵偡丅
仸丗僼僃儞僞僯儖揬晅嵻偵偮偄偰偼揧晅暥彂偺姺嶼昞傪嶲徠丅12.5兪g/h 偵憡摉偡傞丅
亂嶲峫暥專亃
1乯 National Comprehensive Cancer Network乮Version 1. 2009乯丗NCCN Clinical Practice Guidelines in Oncology. Adult cancer pain.
2乯 Hanks GW, de Conno F, Cherny N, et al. Morphine and alternative opioids in cancer pain丟the EAPC recommendations. Br J Cancer 2001丟84丗587-93
3乯 Fine PG, Portenoy RK. Establishing乬best practices乭for opioid rotation丗conclusions of an expert panel. J Pain Symptom Manage. 2009丟38丗418-25
俇丏奺僆僺僆僀僪偺栻棟妛揑摿挜乮昞4乯
❶ 杻栻惈捔捝栻
1 乯僐僨僀儞
乵嶌梡婡彉乶丂僐僨僀儞偺僆僺僆僀僪庴梕懱偵懳偡傞恊榓惈偼掅偔丆偦偺捔捝岠壥偼僐僨僀儞偺堦晹偑O-扙儊僠儖壔偝傟偨儌儖僸僱偵傛傞傕偺偱偁傞丅
乵媧廂丒戙幱丒攔煏乶丂宱岥惢嵻偼娞弶夞捠夁岠壥偑彮側偔丆栺0.8 帪娫偱嵟崅寣拞擹搙偵摓払偡傞丅僐僨僀儞偺僆僺僆僀僪庴梕懱傊偺恊榓惈偼掅偄偑丆僐僨僀儞偑娞憻偱戙幱偝傟傞偲丆栺10亾偑僠僩僋儘儉P450仏偺CYP2D6 偵傛傝儌儖僸僱偲側傝丆捔捝岠壥傪傕偨傜偡丅擔杮恖偺栺20乣40亾偼CYP2D6妶惈偑掅偔乮poor metabolizer 傕偟偔偼intermediate metabolizer乯丆儌儖僸僱偑惗惉偝傟偵偔偄偨傔丆僐僨僀儞偺捔捝岠壥偼敪婗偝傟偵偔偄乮昞5乯丅
乵摿丂挜乶丂僐僨僀儞偼捔奝嶌梡傪桳偟丆偙傟偼僐僨僀儞偦偺傕偺偺嶌梡偱偁傞丅WHO 偺暘椶偱偼庛僆僺僆僀僪偵暘椶偝傟丆拞摍搙傑偱偺捝傒偺帯椕偵巊梡偝傟丆儌儖僸僱偺1/6乣1/10 偺捔捝嶌梡傪桳偟偰偄傞丅暃嶌梡偲偟偰丆庡偵埆怱丒歲揻丆曋旈偍傛傃柊婥偑偁傞丅
仏丗僠僩僋儘儉P450
傎偲傫偳偡傋偰偺惗暔偵懚嵼偡傞巁壔峺慺丅僸僩偱偼尰嵼栺50 庬偑曬崘偝傟丆CYP3A4丆CYP2A6乮CYP亖cytochrome P450乯側偳偑偁傞丅娞憻偵懡偔懚嵼偟丆栻暔戙幱偺庡梫側峺慺丅
| 僆僺僆僀僪 | 兪庴梕懱 | 兟庴梕懱 | 內庴梕懱 |
|---|---|---|---|
僐僨僀儞 |
亄 | ||
僩儔儅僪乕儖 |
亄仸 | ||
儌儖僸僱 |
亄亄亄 | 亄 | |
僆僉僔僐僪儞 |
亄亄亄 | ||
僼僃儞僞僯儖 |
亄亄亄 | ||
儊僒僪儞 |
亄亄亄 | ||
僞儁儞僞僪乕儖 |
亄 | ||
儁儞僞僝僔儞 |
亄亄乮P乯 | 亄 | 亄亄 |
僽僾儗僲儖僼傿儞 |
亄亄亄乮P乯 | 亄亄乮P乯 | 亄亄亄乮P乯 |
乮P乯偼晹暘嶌摦栻偱偁傞偙偲傪帵偡丅
仸丗僩儔儅僪乕儖帺懱偵寢崌恊榓惈偼側偔丆戙幱暔偑晹暘嶌摦栻偲偟偰嶌梡偡傞丅
| 僆僺僆僀僪 | 庡側 戙幱晹埵 |
枹曄壔懱擜拞 攔煏棪 乮恡攔煏棪乯 |
暔幙偲偟偰偺 敿尭婜 |
庡側 戙幱宱楬 |
戙幱暔 乮捔捝妶惈偺桳柍乯 |
|---|---|---|---|---|---|
僐僨僀儞 |
娞憻 | 栺3乣16亾 | 栺2.5乣3.5 帪娫 | CYP2D6 |
儌儖僸僱乮桳乯 |
僩儔儅僪乕儖 |
娞憻 | 栺30亾 | 栺6 帪娫 | CYP2D6 |
O-僨僗儊僠儖僩 |
CYP3A4 |
N-僨僗儊僠儖僩 |
||||
儌儖僸僱 |
娞憻 | 栺8乣10亾 | 栺2乣4 帪娫 | 僌儖僋儘儞 |
M6G乮桳乯 |
僌儖僋儘儞 |
M3G仸 |
||||
僆僉僔僐僪儞 |
娞憻 | 栺5.5乣19亾 | 栺3.5乣4 帪娫 | CYP3A4 |
僲儖僆僉僔僐僪儞 |
CYP2D6 |
僆僉僔儌儖僼僅儞 |
||||
僼僃儞僞僯儖 |
娞憻 | 栺10亾 | 栺4 帪娫 | CYP3A4 |
僲儖僼僃儞僞僯儖 |
儊僒僪儞 |
娞憻 | 栺21亾 | 栺30乣40 帪娫 | CYP3A4丆 |
EDDP乮柍乯 |
僞儁儞僞僪乕儖 |
娞憻 | 栺3亾 | 栺4乣5 帪娫 | 僌儖僋儘儞 |
僞儁儞僞僪乕儖 |
儁儞僞僝僔儞 |
娞憻 | 栺5乣8亾 | 栺2乣3 帪娫 | 僌儖僋儘儞 |
儁儞僞僝僔儞僌儖 |
僽僾儗僲儖僼傿儞 |
娞憻 | 栺1亾 | 栺2 帪娫 | CYP3A4 |
僲儖僽僾儗僲儖 |
仸丗捔捝妶惈偼側偄偑恄宱撆惈傪桳偟偰偄傞偲偺曬崘傕偁傞丅
2 乯僩儔儅僪乕儖
乵嶌梡婡彉乶丂僩儔儅僪乕儖偼僐僨僀儞椶帡偺崌惉壔崌暔偱偁傝丆偦偺捔捝岠壥偼丆兪僆僺僆僀僪庴梕懱偵懳偡傞庛偄恊榓惈偲僙儘僩僯儞丒僲儖傾僪儗僫儕儞嵞庢傝崬傒慾奞嶌梡傪偁傢偣傕偮偙偲偱敪婗偝傟傞偲峫偊傜傟偰偄傞丅僩儔儅僪乕儖偺戙幱暔偱偁傞儌僲-O-扙儊僠儖懱偼丆兪僆僺僆僀僪庴梕懱偵懳偟偰枹曄壔懱傛傝傕崅偄恊榓惈傪桳偡傞偨傔丆僩儔儅僪乕儖偺捔捝嶌梡偺堦晹偵婑梌偡傞偲峫偊傜傟偰偄傞丅
乵媧廂丒戙幱丒攔煏乶丂僩儔儅僪乕儖宱岥惢嵻偺惗懱撪棙梡棪仏偼栺75亾偱偁傝丆拞悤堏峴惈傕椙岲偱偁傞丅庡偵娞憻僠僩僋儘儉P450 偺CYP2D6 偍傛傃CYP3A4 偱戙幱偝傟丆O-僨僗儊僠儖僩儔儅僪乕儖偍傛傃N-僨僗儊僠儖僩儔儅僪乕儖偵曄姺偝傟丆恡傛傝僩儔儅僪乕儖偲偟偰栺30亾丆戙幱暔偲偟偰栺60亾偑攔煏偝傟傞丅O-僨僗儊僠儖僩儔儅僪乕儖偼丆兪僆僺僆僀僪庴梕懱偵嶌梡偟僩儔儅僪乕儖偺悢攞偺捔捝岠壥傪敪婗偡傞乮昞5乯丅
乵摿丂挜乶丂僩儔儅僪乕儖偼丆WHO 曽幃偑傫醬捝帯椕朄偺戞擇抜奒栻孮偵暘椶偝傟偰偄傞丅嶌梡敪尰帪娫偍傛傃帩懕帪娫偼儌儖僸僱偲摨掱搙偱偁傞丅僩儔儅僪乕儖偼偦偺嶌梡婡彉偐傜恄宱忈奞惈醬捝偵岠壥揑偱偁傞偙偲偑曬崘偝傟偰偄傞丅曋旈丆埆怱丒歲揻偺敪惗昿搙偼掅偄丅偗偄傟傫敪嶌傪堷偒婲偙偡偙偲偑偁傞丅
仏丗惗懱撪棙梡棪
搳梌偟偨栻暔偺壗亾偑惗懱撪乮寣拞乯偵庢傝崬傑傟丆柍懯側偔妶梡偝傟傞偐偲偄偆栻暔偺棙梡棪乮媧廂棪乯丅惗暔妛揑棙梡棪丆僶僀僆傾儀僀儔價儕僥傿乮bioavailability乯偲傕偄偆丅
3 乯儌儖僸僱
乵嶌梡婡彉乶丂戙昞揑側僆僺僆僀僪偱偁傞儌儖僸僱偼丆兪僆僺僆僀僪庴梕懱偵懳偡傞慖戰惈偑斾妑揑崅偔乮兟丆內僆僺僆僀僪庴梕懱傛傝傕悢攞乣悢廫攞乯丆偦偺嶌梡偺傎偲傫偳偑兪僆僺僆僀僪庴梕懱傪夘偟偰敪尰偡傞丅
乵媧廂丒戙幱丒攔煏乶丂宱岥搳梌偝傟偨儌儖僸僱偼丆堓挵娗偐傜媧廂偝傟傞丅懍曻惈宱岥惢嵻偼丆栺0.5乣1.3 帪娫偱嵟崅寣拞擹搙偵摓払偡傞丅傑偨丆彊曻惈宱岥惢嵻偼丆栺1.9乣7.3 帪娫偱嵟崅寣拞擹搙偵摓払偡傞丅媧廂偝傟偨儌儖僸僱偼娞弶夞捠夁岠壥偵傛傝戙幱偝傟丆惗懱撪棙梡棪偼19乣47亾乮暯嬒25亾乯偱偁傞丅慡恎弞娐偵摓払偟偨儌儖僸僱偼丆僌儖僋儘儞巁書崌偵傛傝丆栺44乣55亾偑儌儖僸僱-3-僌儖僋儘僯僪乮M3G乯偵丆栺9乣10亾偑儌儖僸僱-6-僌儖僋儘僯僪乮M6G乯偵戙幱偝傟丆8乣10亾偑枹曄壔懱乮儌儖僸僱乯偲偟偰擜拞偐傜攔煏偝傟傞丅M6G 偍傛傃M3G 偼丆傎偲傫偳恡憻偐傜攔煏偝傟傞乮昞5乯丅
乵摿丂挜乶丂儌儖僸僱偼丆懡偔偺偑傫醬捝娚榓僈僀僪儔僀儞偵偍偄偰丆朙晉側巊梡宱尡側偳偐傜戞堦慖戰栻偲偟偰悇彠偝傟偰偒偨丅傑偨丆宱岥傗惷柆撪丆捈挵撪丆旂壓丆峝枌奜丆僋儌枌壓峯撪傊搳梌偱偒傞丅儌儖僸僱偺戙幱暔偱偁傞M6G 偼嫮椡側捔捝嶌梡傪桳偟偰偍傝丆傑偨丆擼堏峴惈偑儌儖僸僱傛傝傕掅偔丆備偭偔傝偲寣塼擼娭栧傪捠夁偡傞偨傔偵丆嶌梡帩懕帪娫偑挿偄丅堦曽丆傕偆堦偮偺戙幱暔偱偁傞M3G 偼丆僆僺僆僀僪庴梕懱偵懳偟偰傎偲傫偳恊榓惈傪傕偨偢丆捔捝嶌梡偼帵偝側偄偑丆偑傫醬捝姵幰傊儌儖僸僱傪戝検搳梌偟偨嵺偵擣傔傜傟傞捝妎夁晀仏1 傗傾儘僨傿僯傾仏2 偺敪尰偵娭梌偟偰偄傞壜擻惈偑帵嵈偝傟偰偄傞丅庡側暃嶌梡偲偟偰丆埆怱丒歲揻丆曋旈偍傛傃柊婥偑偁傞丅
仏1丗捝妎夁晀乮hyperalgesia乯
捝妎偵懳偡傞姶庴惈偑槾恑偟偨忬懺丅捠忢偱偼捝傒傪姶偠側偄掱搙偺捝傒偺巋寖偵懳偟偰捝傒傪姶偠傞偙偲丅
乮嶲峫乯捝妎撦杻乮hypoalgesia乯
捝妎偵懳偡傞姶庴惈偑掅壓偟偨忬懺丅捠忢偱偼捝傒傪惗偠傞巋寖偵懳偟偰捝傒傪姶偠側偄丒姶偠偵偔偄偙偲丅
仏2丗傾儘僨傿僯傾乮allodynia乯
捠忢偱偼捝傒傪婲偙偝側偄巋寖乮乽怗傞乿側偳乯偵傛偭偰堷偒婲偙偝傟傞捝傒丅堎捝乮徢乯偲栿偝傟傞応崌偑偁傞偑丆杮僈僀僪儔僀儞偱偼丆傾儘僨傿僯傾偲昞尰偟偨丅
4 乯僆僉僔僐僪儞
乵嶌梡婡彉乶丂僆僉僔僐僪儞偼丆敿崌惉僥僶僀儞桿摫懱偱偁傝丆嫮僆僺僆僀僪偵暘椶偝傟傞丅偦偺栻棟嶌梡偼庡偵兪僆僺僆僀僪庴梕懱傪夘偟偰敪尰偡傞丅
乵媧廂丒戙幱丒攔煏乶丂懍曻惈宱岥惢嵻偼栺1.7乣1.9 帪娫偱嵟崅寣拞擹搙偵摓払偡傞丅傑偨丆彊曻惈宱岥惢嵻偼栺4.0 帪娫偱嵟崅寣拞擹搙偵摓払偡傞丅宱岥僆僉僔僐僪儞偺惗懱撪棙梡棪偼栺60亾乮50乣87亾乯偱偁傞丅僠僩僋儘儉P450 偺CYP2D6 偍傛傃CYP3A4 偵傛傝丆僲儖僆僉僔僐僪儞偍傛傃僆僉僔儌儖僼僅儞偵戙幱偝傟傞丅僲儖僆僉僔僐僪儞偼丆庡戙幱暔偱偁傞偑丆旕妶惈戙幱暔偱偁傞丅傑偨丆僆僉僔儌儖僼僅儞偼捔捝妶惈傪帵偡偑丆偦偺AUC仏3乲栻暔寣拞擹搙乮帪娫乯嬋慄壓柺愊乴偼丆僆僉僔僐僪儞AUC 偺栺1.4亾偲偛偔旝検偱偁傞丅僆僉僔僐僪儞偼傎偲傫偳偑娞憻偱戙幱偝傟傞偑丆栺5.5乣19亾偑枹曄壔懱偲偟偰擜拞偐傜攔煏偝傟傞乮昞5乯丅
乵摿丂挜乶丂僆僉僔僐僪儞偼丆宱岥丆惷柆撪偍傛傃旂壓傊搳梌偡傞偙偲偑偱偒傞丅傑偨丆惷柆撪搳梌偵偍偗傞儌儖僸僱偲僆僉僔僐僪儞偺捔捝椡壙偺斾偼栺2丗3 偱偁傞丅宱岥搳梌帪偼丆僆僉僔僐僪儞偺惗懱撪棙梡棪偑儌儖僸僱偺栺2 攞偱偁傞偨傔丆儌儖僸僱偲僆僉僔僐僪儞偺捔捝椡壙偺斾偼栺3丗2 偲側傞丅庡側暃嶌梡偲偟偰丆埆怱丒歲揻丆曋旈偍傛傃柊婥偑偁傝丆儌儖僸僱偲傎傏摨摍偱偁傞丅
仏3丗AUC乮area under the drug concentration time curve乯
栻暔寣拞擹搙乮帪娫乯嬋慄壓柺愊丅栻暔寣拞擹搙傪宱帪揑偵昞偟偨嬋慄僌儔僼偲帪娫幉乮墶幉乯偵埻傑傟偨晹暘偺柺愊丅寣拞偵庢傝崬傑傟偨栻偺検乮媧廂棪乯偺巜昗偲偟偰梡偄傞丅
5 乯僼僃儞僞僯儖
乵嶌梡婡彉乶丂僼僃儞僞僯儖偼丆僼僃僯儖僺儁儕僕儞娭楢偺崌惉僆僺僆僀僪偱偁傝丆杻悓曗彆栻偲偟偰巊梡偝傟偰偒偨丅兪僆僺僆僀僪庴梕懱偵懳偡傞慖戰惈偑旕忢偵崅偔丆姰慡嶌摦栻偲偟偰嶌梡偡傞丅僼僃儞僞僯儖偺捔捝岠壥偼丆儌儖僸僱偲椶帡偟偰偍傝丆惷柆撪搳梌偟偨応崌丆僼僃儞僞僯儖偺捔捝嶌梡偼儌儖僸僱偺栺50乣100 攞偱偁傞丅
乵媧廂丒戙幱丒攔煏乶丂宱旂媧廂宆惢嵻乮僼僃儞僞僯儖揬晅嵻乯偺惗懱撪棙梡棪偼寁嶼忋57乣146亾乮暯嬒92亾乯偱偁傞丅弶夞揬晅屻1乣2 帪娫偱寣拞偵僼僃儞僞僯儖偼専弌偝傟丆17乣48 帪娫偱嵟崅寣拞擹搙偵摓払偡傞丅揬晅2 夞栚埲崀偵掕忢忬懺偵摓払偡傞丅傑偨丆宱岥峯擲枌媧廂宆惢嵻乮僼僃儞僞僯儖岥峯擲枌媧廂嵻乯偼丆僆僺僆僀僪懍曻惈宱岥惢嵻偵斾傋媧廂偑憗偄丅僼僃儞僞僯儖偼傎偲傫偳偑娞憻偱戙幱偝傟丆庡偵僠僩僋儘儉P450偺CYP3A4 偵傛傝丆僲儖僼僃儞僞僯儖偵戙幱偝傟傞丅僲儖僼僃儞僞僯儖偼旕妶惈戙幱暔偱偁傞丅僼僃儞僞僯儖偼帀梟惈偑崅偔丆寣塼擼娭栧傪懍傗偐偵堏峴偡傞乮昞5乯丅
乵摿丂挜乶丂僼僃儞僞僯儖偼丆宱旂丆宱岥峯擲枌丆惷柆撪丆旂壓丆峝枌奜丆僋儌枌壓峯撪傊搳梌偡傞偙偲偑偱偒傞丅惷柆撪搳梌偟偨僼僃儞僞僯儖偑嵟戝捔捝岠壥偵払偡傞帪娫偼栺5 暘偲儌儖僸僱傗懠偺僆僺僆僀僪偲斾妑偟偰懍岠惈偑偁傞丅帀梟惈偑崅偔斾妑揑暘巕検偑彫偝偄偨傔丆旂晢媧廂偑椙岲偱偁傝丆揬晅嵻偲偟偰傕巊梡偝傟偰偄傞丅傑偨丆岥峯擲枌媧廂嵻偼僆僺僆僀僪懍曻惈宱岥惢嵻傛傝媧廂偑憗偄偨傔丆傛傝懄岠惈偑偁傞丅暃嶌梡偲偟偰丆儌儖僸僱偲摨條偵丆埆怱丒歲揻偑偁傞偑丆曋旈偍傛傃柊婥偼斾妑揑彮側偄丅
6 乯儊僒僪儞仸
乵嶌梡婡彉乶丂儊僒僪儞偼丆崌惉僕僼僃僯儖僿僾僞儞桿摫懱偱偁傝丆偦偺捔捝岠壥偼丆兪僆僺僆僀僪庴梕懱偵懳偡傞恊榓惈偲NMDA 庴梕懱漢峈嶌梡偵傛傝敪婗偡傞偲峫偊傜傟傞丅
乵媧廂丒戙幱丒攔煏乶丂儊僒僪儞宱岥惢嵻偺惗懱撪棙梡棪偼栺85亾偱丆拞悤堏峴惈傕椙岲偱偁傞丅栻岠敪尰帪娫偼栺30 暘偲斾妑揑憗偄丅傑偨丆嶌梡帩懕帪娫偼扨夞搳梌偱4乣5 帪娫丆斀暅搳梌偱8乣12 帪娫掱搙偱偁傞丅庡偵娞憻僠僩僋儘儉P450 偺CYP3A4 偍傛傃CYP2B6 偱戙幱偝傟丆EDDP乮2-ethylidene-1,5-dimethyl-3,3-diphenylpyrrolidine乯偵曄姺偝傟傞丅戙幱暔偵偼妶惈偼側偄丅儊僒僪儞偼傎偲傫偳娞憻偱戙幱偝傟傞偑丆栺21亾偑枹曄壔懱偲偟偰擜拞偐傜攔煏偝傟傞丅
乵摿丂挜乶丂儊僒僪儞偼岝妛堎惈懱傪桳偟丆兪庴梕懱偺寢崌恊榓惈偼d 懱傛傝傕l 懱偱栺10 攞崅偄丅NMDA 庴梕懱慾奞嶌梡偼d 懱偲l 懱偱傎傏摨摍偱偁傞丅徚幐敿尭婜偑栺30乣40 帪娫偲挿偄偨傔丆搳梌屻彊乆偵寣拞擹搙偼忋徃偟丆掕忢忬懺偵払偡傞傑偱偵栺1 廡娫傪梫偡傞丅傑偨丆傾儖僇儕擜偱儊僒僪儞偺恡攔煏偑抶墑偟偨傝丆帺屓峺慺桿摫傪婲偙偡偙偲傕曬崘偝傟丆寣拞擹搙傪梊應偡傞偙偲偼崲擄偱偁傞丅暃嶌梡偲偟偰丆QT 墑挿偍傛傃屇媧梷惂偺曬崘偑懡偔丆偦偺巊梡偵偁偨偭偰偼廫暘側拲堄偑昁梫偱偁傞丅
仏丗杮朚偱2012 擭9 寧偵惢憿彸擣偝傟丆2013 擭3 寧偐傜敪攧偝傟偨儊僒僪儞偼丆懠偺嫮僆僺僆僀僪偱帯椕崲擄側拞摍搙偐傜崅搙偺醬捝傪敽偆奺庬偑傫偵偍偗傞捔捝岠壥偑婜懸偝傟傞丅偟偐偟丆挷愡偺擄偟偝側偳偐傜偦偺巊梡偵嵺偟偰偼丆堛巘偼偑傫醬捝偺娗棟偵惛捠偟偰偄傞偩偗偱偼側偔惢憿斕攧嬈幰偺採嫙偡傞島廗傪庴島偡傞偙偲丆栻嵻巘偼島廗傪庴島偟偨堛巘偱偁傞偙偲傪妋擣偡傞偙偲側偳偑捠払偱帵偝傟偰偄傞丅
7 乯僞儁儞僞僪乕儖
乵嶌梡婡彉乶丂僞儁儞僞僪乕儖偺捔捝嶌梡偼丆庡偲偟偰僆僺僆僀僪兪庴梕懱嶌摦嶌梡偍傛傃愐悜屻妏偵偍偗傞僲儖傾僪儗僫儕儞嵞庢傝崬傒慾奞嶌梡偵婎偯偔偲峫偊傜傟偰偄傞丅
乵媧廂丆戙幱丆攔煏乶丂彊曻惈宱岥惢嵻偺惗懱撪棙梡棪偼栺32亾偱偁傞丅寣燋抈敀寢崌棪偼栺20亾偱偁傝丆徚幐敿尭婜偼栺4乣5 帪娫偱偁傞丅娞憻偱庡偵僌儖僋儘儞巁書崌偵傛傝戙幱偝傟丆妶惈偺側偄僞儁儞僞僪乕儖-O-僌儖僋儘僯僪偲側傞丅僞儁儞僞僪乕儖偼娞憻偱戙幱偝傟偨屻丆傎偲傫偳偑擜拞偵攔煏偝傟丆栺3亾偑枹曄壔懱偱偁傞丅
乵摿丂挜乶丂彊曻惈宱岥惢嵻偼TRF乮Tamper Resistant Formulation丗夵曄杊巭惢嵻乯偱旕忢偵峝偔丆婡夿揑乮姎傓丆偡傝偮傇偡乯偍傛傃壔妛揑乮悈傗偦偺懠偺梟攠梟偐偡乯偵夵偞傫偡傞偙偲偑偱偒側偄偨傔丆栻暔棎梡傪杊巭偡傞偙偲偑偱偒傞丅摍捔捝梡検斾偼僞儁儞僞僪乕儖宱岥丗儌儖僸僱宱岥丗僆僉僔僐僪儞宱岥亖100丗30丗20乮mg/擔乯偱偁傞丅
❷ 杻栻漢峈惈捔捝栻
僆僺僆僀僪嶌摦栻偑懚嵼偟側偄忬嫷偱偼嶌摦栻偲偟偰嶌梡偡傞偑丆僆僺僆僀僪嶌摦栻偺懚嵼壓偱偼偦偺嶌梡偵漢峈偡傞嶌梡傪傕偮捔捝栻丅
1 乯儁儞僞僝僔儞
乵嶌梡婡彉乶丂儁儞僞僝僔儞偼內僆僺僆僀僪庴梕懱偵懳偟偰嶌摦栻偲偟偰嶌梡偟丆兪僆僺僆僀僪庴梕懱偵懳偟偰偼漢峈栻仏1 傕偟偔偼晹暘嶌摦栻仏2 偲偟偰嶌梡偡傞丅儁儞僞僝僔儞偼捔捝丆捔惷丆屇媧梷惂傪娷傔儌儖僸僱側偳偺僆僺僆僀僪偲傎傏椶帡偡傞嶌梡傪帵偡丅偦偺捔捝嶌梡偼庡偵內僆僺僆僀僪庴梕懱傪夘偟偰敪尰偡傞偑丆堦晹兪僆僺僆僀僪庴梕懱傕夘偟偰偄傞丅傑偨丆捔捝嶌梡偺揤堜岠壥仏3 傪桳偡傞丅
乵媧廂丒戙幱丒攔煏乶丂宱岥惢嵻偼栺2.0 帪娫偱嵟崅寣拞擹搙偵摓払偡傞丅枹曄壔懱偱恡傛傝攔煏偝傟傞儁儞僞僝僔儞偼5乣8亾偱偁傞偨傔丆傎偲傫偳偑娞憻偱戙幱偝傟丆庡側戙幱宱楬偼僌儖僋儘儞巁偲偺書崌偱偁傞丅戙幱暔偵偼妶惈偼懚嵼偟側偄乮昞5乯丅
乵摿丂挜乶丂儌儖僸僱傪挿婜娫搳梌偝傟偰偄傞姵幰偵懳偟偰丆儁儞僞僝僔儞傪搳梌偡傞偲兪僆僺僆僀僪庴梕懱漢峈嶌梡偵傛傝棧扙徢岓仏4 傗捔捝岠壥掅壓傪堷偒婲偙偡壜擻惈偑偁傞丅歲揻偼儌儖僸僱傎偳傒傜傟側偄偑丆晄埨丆尪妎側偳偺惛恄徢忬偑敪尰偡傞偙偲偑偁傞丅
仏1丗漢峈栻
庴梕懱偵嶌梡偟偰丆懠偺惗懱撪暔幙側偳偑庴梕懱偵寢崌偡傞偙偲傪朩偘傞栻暔丅漢峈栻帺懱偼庴梕懱傪妶惈壔偡傞嶌梡傪傕偨偢丆惗懱墳摎傪婲偙偝側偄丅幷抐栻丆傾儞僞僑僯僗僩偲傕偄偆丅
仏2丗晹暘嶌摦栻
庴梕懱偲寢崌偟偰丆庴梕懱傪妶惈忬懺偵偡傞栻嵻傪嶌摦栻乮傾僑僯僗僩乯偲偄偄丆偙偺偆偪庴梕懱偵寢崌偡傞偑丆100亾偺妶惈壔傪堷偒婲偙偝側偄栻丅
仏3丗揤堜岠壥乮ceiling effect乯
偁傞掱搙偺検埲忋丆搳梌検傪憹傗偟偰傕捔捝岠壥偑摢懪偪偵側傞偙偲丅桳岠尷奅偲傕偄偆丅
仏4丗棧扙徢岓丒棧扙徢岓孮
椪彴偱偼栻暔偺撍慠偺媥栻偵傛傞恎懱徢忬傪棧扙徢岓孮乮withdrawal syndrome乯偲昞尰偡傞偙偲偑堦斒揑偱偁傞丅戅栻徢忬丆戅栻挜岓偲傕偄傢傟傞偑丆杮僈僀僪儔僀儞偵偍偄偰偼丆僈僀僪儔僀儞傪巊梡偡傞堛椕廬帠幰偺崿棎傪旔偗傞偨傔丆杮暥傪捠偟偰棧扙徢岓丒棧扙徢岓孮偵摑堦偟偰巊梡偡傞丅
2 乯僽僾儗僲儖僼傿儞
乵嶌梡婡彉乶丂僽僾儗僲儖僼傿儞偼兪僆僺僆僀僪庴梕懱偵懳偟偰嶌摦栻偲偟偰嶌梡偟丆內僆僺僆僀僪庴梕懱偵懳偟偰偼漢峈嶌梡傪帵偡丅儌儖僸僱傛傝25乣50 攞嫮偄岠椡傪傕偪丆儌儖僸僱偲椶帡偡傞嶌梡傪帵偡偑丆揤堜岠壥傪桳偡傞丅僽僾儗僲儖僼傿儞偼丆僆僺僆僀僪庴梕懱偵懳偟偰恊榓惈偑崅偔丆偐偮崅偄帀梟惈傪傕偮偨傔丆庴梕懱偐傜偺夝棧偑娚傗偐偱偁傝丆挿帪娫偺嶌梡乮栺6乣9 帪娫乯傪帵偡丅
乵媧廂丒戙幱丒攔煏乶丂嵖嵻偼栺1.0乣2.0 帪娫偱嵟崅寣拞擹搙偵摓払偡傞丅僽僾儗僲儖僼傿儞偼庡偵娞憻偱戙幱偝傟丆僠僩僋儘儉P450 偺CYP3A4 偵傛傝僲儖僽僾儗僲儖僼傿儞偵戙幱偝傟傞乮昞5乯丅
乵摿丂挜乶丂僽僾儗僲儖僼傿儞偼捈挵撪丆惷柆撪丆旂壓傊搳梌偡傞偙偲偑偱偒傞丅拲幩偵偍偄偰2 mg/擔偱揤堜岠壥偑傒傜傟傞偨傔丆嫮僆僺僆僀僪偵曄峏偡傞昁梫偑偁傞丅僽僾儗僲儖僼傿儞偼丆兪僆僺僆僀僪庴梕懱偵懳偡傞恊榓惈偑儌儖僸僱傛傝傕嫮偄偨傔丆戝検偵儌儖僸僱傪搳梌偟偰偄傞姵幰偵僽僾儗僲儖僼傿儞傪搳梌偡傞偲丆兪僆僺僆僀僪庴梕懱偵寢崌偱偒傞儌儖僸僱偲嫞崌偡傞偨傔偵丆憤崌揑偵捔捝岠壥偑庛傑傞壜擻惈偑偁傞丅庡側暃嶌梡偲偟偰丆埆怱丒歲揻丆曋旈偍傛傃柊婥偑偁傞丅
俈丏摿庩側昦懺偱偺僆僺僆僀僪偺慖戰乯
❶ 恡婡擻忈奞
儌儖僸僱偼丆娞憻偱庡偵僌儖僋儘儞巁書崌偝傟丆M3G 偲M6G 偵曄姺偝傟傞丅M6G 偼捔捝偍傛傃捔惷嶌梡傪帵偡偙偲偑抦傜傟偰偄傞丅M3G 偲M6G 偼傎偲傫偳恡偐傜攔煏偝傟傞偨傔丆恡婡擻忈奞姵幰偵儌儖僸僱傪巊梡偡傞偲M3G 偍傛傃M6G 偑拁愊偟丆捔惷側偳偺暃嶌梡傊偺懳張偑崲擄偵側傞丅偦偺偨傔丆恡婡擻忈奞姵幰偵偼儌儖僸僱傪巊梡偟側偄傎偆偑朷傑偟偄丅巊梡偡傞嵺偼尭検偁傞偄偼搳梌娫妘傪墑挿偡傞丅摿偵丆崅搙側恡婡擻忈奞傪桳偡傞姵幰偱偼儌儖僸僱傪巊梡偡傋偒偱偼側偄丅
摨條偵僐僨僀儞偼10亾掱搙偑儌儖僸僱偵曄姺偝傟丆偝傜偵M3G 偍傛傃M6G 偵曄姺偝傟傞偨傔丆恡婡擻忈奞姵幰偵僐僨僀儞傪巊梡偟側偄偙偲偑朷傑偟偄丅巊梡偡傞嵺偼尭検偁傞偄偼搳梌娫妘傪墑挿偡傞丅
僆僉僔僐僪儞偼丆娞憻偱戙幱偝傟庡偵僲儖僆僉僔僐僪儞偍傛傃僆僉僔儌儖僼僅儞偵曄姺偝傟傞丅僆僉僔儌儖僼僅儞偼捔捝妶惈傪桳偡傞偑偛偔彮検偟偐惗惉偝傟側偄丅傑偨丆巊梡偡傞嵺偼廫暘偵拲堄偟偰怲廳側娤嶡偑昁梫偱偁傞丅
僼僃儞僞僯儖偼丆娞憻偱庡偵旕妶惈戙幱暔偱偁傞僲儖僼僃儞僞僯儖偵曄姺偝傟傞丅椪彴宱尡偐傜斾妑揑埨慡偵恡婡擻忈奞姵幰偵巊梡偱偒傞偑丆寣拞擹搙偑忋徃偡傞偨傔尭検偟偰巊梡偡傞丅挿婜娫偵媦傇嵺偼岠壥偍傛傃暃嶌梡傪拲堄怺偔娤嶡偡傞昁梫偑偁傞丅
儊僒僪儞偼丆娞憻偱庡偵旕妶惈戙幱暔偱偁傞EDDP 偵曄姺偝傟傞丅斾妑揑埨慡偵巊梡偱偒傞偑丆恡攔煏偑栺20亾偁傝寣拞擹搙偑忋徃偡傞偲峫偊傜傟傞偨傔尭検偟偰巊梡偡傞丅巊梡偡傞嵺偼廫暘偵拲堄偟偰怲廳側娤嶡偑昁梫偱偁傞丅
❷ 摟丂愅
儌儖僸僱偍傛傃偦偺戙幱暔偱偁傞M3G丆M6G 偼丆寣塼摟愅帪偵寣塼拞偐傜堦晹彍嫀偝傟傞偑丆寣塼摟愅屻偵拞悤恄宱宯偲寣燋偲偺娫偱嵞傃暯峵忬懺偲側傞丅偦偺偨傔丆旕摟愅帪偵偼M3G 偍傛傃M6G 偑拁愊偡傞丅傑偨丆寣塼摟愅偵傛傞堦帪揑側寣拞擹搙掅壓偵傛傝丆摟愅拞偁傞偄偼摟愅屻偵僆僺僆僀僪偺捛壛搳梌偑昁梫偵側傞壜擻惈偑偁傞丅偟偨偑偭偰丆摟愅姵幰偵偼儌儖僸僱傪巊梡偟側偄傎偆偑朷傑偟偄丅
摨條偵僐僨僀儞偼慜弎偺棟桼偱丆摟愅姵幰偵僐僨僀儞傪巊梡偟側偄傎偆偑朷傑偟偄偑丆巊梡偡傞嵺偼尭検偁傞偄偼搳梌娫妘傪墑挿偡傞丅
僆僉僔僐僪儞偼寣塼摟愅帪偺僨乕僞偑朢偟偄丅巊梡偡傞応崌偼尭検偁傞偄偼搳梌娫妘傪墑挿偡傞昁梫偑偁傞丅傑偨丆儌儖僸僱摨條偵抈敀寢崌棪偑掅偄偨傔寣塼拞偐傜堦晹彍嫀偝傟丆堦帪揑側寣拞擹搙掅壓偵傛傝丆摟愅拞偁傞偄偼摟愅屻偵僆僺僆僀僪偺捛壛搳梌偑昁梫偵側傞壜擻惈偑偁傞丅
僼僃儞僞僯儖偼丆搳梌検偺挷愡側偟偵斾妑揑埨慡偵摟愅姵幰偵巊梡偱偒傞丅抈敀寢崌棪仏1 偑崅偔摟愅枌偵媧拝偡傞偙偲偑偁傞偨傔丆醬捝偺娚榓偑崲擄偵側傞応崌偼僆僺僆僀僪僗僀僢僠儞僌傪専摙偡傞丅傑偨丆挿婜娫偵媦傇嵺偼拲堄怺偔姵幰傪娤嶡偡傞昁梫偑偁傞丅
儊僒僪儞偼丆暘晍梕愊偑戝偒偔丆抈敀寢崌棪偑崅偄偨傔丆摟愅偱彍嫀偝傟偵偔偄偲峫偊傜傟傞丅儊僒僪儞偼戙幱暔偵妶惈偑側偔丆摟愅拞傕傎偲傫偳彍嫀偝傟側偄偺偱摟愅姵幰偵傕斾妑揑埨慡偵巊梡偱偒傞丅巊梡偡傞嵺偼廫暘偵拲堄偟偰怲廳側娤嶡偑昁梫偱偁傞丅
仏1丗抈敀寢崌棪
寣燋抈敀偲寢崌偟偰偄傞栻暔傪寢崌宆栻暔丆寢崌偟偰偄側偄栻暔傪梀棧宆栻暔偲偄偆丅抈敀寢崌棪偲偼憤栻暔検偵懳偡傞寢崌宆偺妱崌偺偙偲丅寢崌宆偼惗懱枌傪捠夁偱偒側偄偨傔丆栻岠偼梀棧宆偺憤検偵傛傝嵍塃偝傟傞丅
❸ 娞婡擻忈奞
儌儖僸僱丆僆僉僔僐僪儞丆僼僃儞僞僯儖丆僐僨僀儞丆儊僒僪儞偼傎偲傫偳偑娞憻偱戙幱偝傟傞偨傔丆娞忈奞帪偵偼戙幱擻偑尭彮偡傞丅偟偨偑偭偰丆娞婡擻忈奞帪偵偼搳梌検偺尭検偁傞偄偼搳梌娫妘傪墑挿偟偰丆栻暔偺拁愊傪杊巭偡傞昁梫偑偁傞丅
乮崙暘廏栫乯
亂嶲峫暥專亃
1乯 Dean M. Opioids in renal failure and dialysis patients. J Pain Symtom Manage 2004丟28丗497-504
2乯 Murtagh FE, Chai MO, Donohoe P, et al. The use of opioid analgesia in end-stage renal disease patients managed without dialysis丗recommendations for practice. J Pain Palliat Care Pharmacother 2007丟21丗5-16
3乯 Frampton JE. Tapentadol immediate release丗a review of its use in the treatment of moderate to severe acute pain. Drugs 2010丟70丗1719-43
俉丏僆僺僆僀僪偵傛傞暃嶌梡偲懳嶔-徚壔婍宯偺暃嶌梡偲懳嶔
儌儖僸僱傪偼偠傔偲偡傞僆僺僆僀僪偵傛傞徚壔婍宯偺庡梫側暃嶌梡偼丆埆怱丒歲揻偲曋旈偱偁傞丅
❶ 埆怱丒歲揻
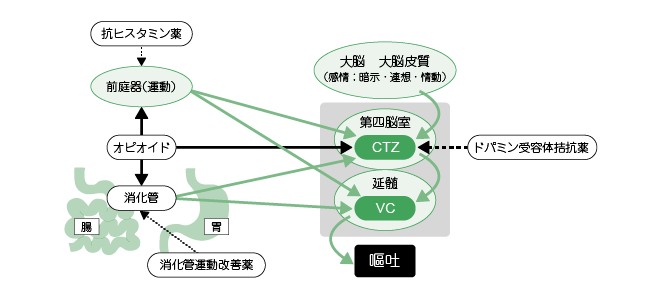
- 埆怱丒歲揻偼丆僆僺僆僀僪偑CTZ乮chemoreceptor trigger zone丗壔妛庴梕婍堷偒嬥懷乯偵朙晉偵敪尰偟偰偄傞兪庴梕懱傪巋寖偡傞偙偲偵傛傝婲偙傞丅妶惈壔偝傟偨兪庴梕懱偑偙偺晹埵偱偺僪僷儈儞仏2 梀棧傪堷偒婲偙偟丆僪僷儈儞D2庴梕懱仏3 偑妶惈壔偝傟丆偦偺寢壥丆歲揻拞悤乮vomiting center丟VC乯偑巋寖偝傟傞偙偲偵傛傞丅傑偨丆慜掚婍偵敪尰偟偰偄傞兪庴梕懱傪巋寖偡傞偙偲偵傛傝僸僗僞儈儞梀棧偑婲偒丆梀棧偝傟偨僸僗僞儈儞偑CTZ 偍傛傃VC 傪巋寖偡傞偙偲偱傕婲偙傞丅偝傜偵偼丆徚壔娗偵偍偄偰丆徚壔娗寰摦塣摦偑梷惂偝傟堓撪梕暔偺掆懾偑婲偙傞偙偲偵傛傝丆媮怱惈偵僔僌僫儖偑揱傢傝CTZ 偍傛傃VC 偑巋寖偝傟傞偙偲偱傕婲偙傞乮恾2乯丅
- 埆怱丒歲揻偼僆僺僆僀僪偺搳梌弶婜偵偟偽偟偽傒傜傟傞暃嶌梡偱偁傞丅
- 捠忢偼僆僺僆僀僪搳梌弶婜丆偁傞偄偼憹検帪偵婲偙傞偙偲偑懡偔丆悢擔埲撪偵懴惈仏4傪 惗偠丆徢忬偑帯傑偭偰偔傞偙偲偑懡偄丅
- 姵幰偵偲偭偰埆怱丒歲揻偼嵟傕晄夣側徢忬偺堦偮偱偁傝丆暈栻傾僪僸傾儔儞僗仏5 傪懝側偆偙偲偵偮側偑傞偙偲傕懡偄偨傔丆愊嬌揑側懳嶔偑昁梫偱偁傞丅
仏2丗僪僷儈儞
擼撪偵懚嵼偡傞恄宱揱払暔幙偺堦偮偱丆夣偺姶忣丆塣摦挷愡丆儂儖儌儞挷愡丆妛廗側偳偵娭傢傞丅傾僪儗僫儕儞丒僲儖傾僪儗僫儕儞偺慜嬱懱丅
仏3丗僪僷儈儞D2庴梕懱
尰嵼丆5 偮偑抦傜傟偰偄傞僪僷儈儞偺庴梕懱偺堦偮丅擼撪偺歲揻拞悤傗丆堓挵偺塣摦傪僐儞僩儘乕儖偡傞恄宱乮暃岎姶恄宱乯偵懚嵼偡傞丅扨偵D2庴梕懱偲傕屇偽傟傞丅
仏4丗懴惈
弶婜偵搳梌偝傟偰偄偨栻暔偺梡検偱摼傜傟偰偄偨栻棟妛揑岠壥偑帪娫宱夁偲偲傕偵尭戅偟丆摨偠岠壥傪摼傞偨傔偵傛傝懡偔偺梡検偑昁梫偵側傞丆恎懱偺栻暔偵懳偡傞惗棟揑弴墳忬懺偱偁傞丅嶲徠丅
仏5丗傾僪僸傾儔儞僗
姵幰偑庡懱偲側偭偰帯椕曽恓偺寛掕偵嶲壛偟丆偦偺寛掕偵廬偭偰帯椕傪庴偗傞偙偲丅廬棃巊傢傟偰偒偨僐儞僾儔僀傾儞僗乮弲庣乯傛傝傕堛椕偺庡懱傪姵幰懁偵抲偄偨峫偊曽丅
乵懳丂嶔乶乮昞6乯丂乮暃嶌梡懳嶔偼Ⅲ-2-1 埆怱丒歲揻偺崁嶲徠乯
- 峈僪僷儈儞嶌梡傪傕偮栻暔乮僾儘僋儘儖儁儔僕儞丆僴儘儁儕僪乕儖側偳乯偑梡偄傜傟傞丅埆怱丒歲揻偑懱摦帪偵婲偙傞応崌傗傔傑偄傪敽偆応崌偼僸僗僞儈儞梀棧傪夘偟偨婡彉偑峫偊傜傟傞偨傔丆峈僸僗僞儈儞栻偺搳梌傪峴偆丅怘屻偵弌尰偡傞埆怱丒歲揻側偳堓撪梕暔挋棷丒挵娗塣摦梷惂偑尨場偲峫偊傜傟傞応崌偼丆徚壔娗塣摦槾恑嶌梡傪傕偮儊僩僋儘僾儔儈僪丆僪儞儁儕僪儞側偳傪搳梌偡傞丅
- 僆僺僆僀僪僗僀僢僠儞僌傪峴偆偙偲偱傕寉夣偡傞偙偲偑偁傞丅僆僺僆僀僪宱岥嵻偐傜揬晅嵻傗拲幩嵻偵搳梌宱楬傪曄偊傞偙偲偱傕寉夣偡傞偙偲偑偁傞丅
- 戞堦慖戰偺惂揻栻偑柍岠偺応崌偼丆旕掕宆峈惛恄昦栻仏乮僆儔儞僓僺儞丆儕僗儁儕僪儞側偳乯偺搳梌偱寉夣偡傞偙偲偑偁傞丅
恾2丂僆僺僆僀僪偵傛傞歲揻偺婡彉
| 庡側嶌梡晹埵 | 栻嵻柤 | 嵻丂宍 | 1 夞搳梌検 |
|---|---|---|---|
CTZ |
僾儘僋儘儖儁儔僕儞 | 忶 |
5 mg |
拲 |
5 mg | ||
| 僴儘儁儕僪乕儖 | 忶 |
0.75 mg | |
拲 |
2.5乣10 mg | ||
慜掚婍 |
僕僼僃儞僸僪儔儈儞/僕僾儘僼傿儕儞 | 忶 |
40 mg/26 mg仸 |
拲 |
2.5乣5 mg | ||
| 僋儘儖僼僃僯儔儈儞儅儗僀儞巁墫 | 忶 |
2 mg | |
拲 |
5 mg | ||
徚壔娗 |
儊僩僋儘僾儔儈僪 | 忶 |
5乣10 mg |
拲 |
10 mg | ||
| 僪儞儁儕僪儞 | 忶 |
5乣10 mg | |
嵖栻 |
60 mg | ||
CTZ丒VC 側偳 |
僆儔儞僓僺儞 | 忶 |
2.5 mg |
| 儕僗儁儕僪儞 | 忶 |
0.5 mg | |
塼 |
0.5 mg |
仸丗僩儔儀儖儈儞® 偲偟偰丅
仏丗旕掕宆峈惛恄昦栻
1980 擭戙屻敿傛傝摫擖偝傟偨怴婯峈惛恄昦栻丅廬棃偺峈惛恄昦栻偲斾妑偟偰丆僪僷儈儞D2庴梕懱埲奜偺恄宱揱払暔幙庴梕懱偵懳偟偰傕慖戰揑偵嶌梡偟丆悕懱奜楬徢忬傪拞怱偲偟偨拞悤恄宱偵懳偡傞暃嶌梡偑彮側偄丅
乮嶲峫乯掕宆峈惛恄昦栻
僪僷儈儞D2庴梕懱偵懳偟偰崅偄恊榓惈傪傕偮漢峈栻偱偁傝丆僴儘儁儕僪乕儖傗僋儘儖僾儘儅僕儞側偳偵戙昞偝傟傞峈惛恄昦栻丅
❷ 曋丂旈
- 曋旈偼僆僺僆僀僪傪搳梌偝傟偨姵幰偵崅昿搙偵婲偙傝丆懴惈宍惉偼傎偲傫偳婲偙傜側偄偨傔壓嵻傪宲懕揑偵搳梌偡傞側偳偺懳嶔偑昁梫偵側傞丅
- 僆僺僆僀僪偼丆奺庬憻婍偐傜偺徚壔峺慺偺暘斿傪梷惂偟丆徚壔娗偺寰摦塣摦傕梷惂偡傞偨傔丆怘暔徚壔偑抶懾偟丆挵娗偱偺怘暔捠夁帪娫偼墑挿偡傞丅偝傜偵怘暔偑戝挵偱挿帪娫偲偳傑傞側偐偱丆悈暘媧廂偼堦抜偲恑傓偨傔曋偼屌偔側傞寢壥丆曋旈偑婲偙傞丅傑偨丆汨栧妵栺嬝偺嬞挘傕崅傑傞偨傔丆攔曋偟偵偔偄忬嫷偲側傞丅
乵懳丂嶔乶乮昞7乯丂乮暃嶌梡懳嶔偼Ⅲ-2-2 曋旈偺崁嶲徠乯
- 僆僺僆僀僪張曽帪偵偼曋旈偑崅昿搙偵擣傔傜傟傞偙偲傪憐掕偟丆壓嵻傪搳梌偡傞側偳偺梊杊揑懳墳偑昁梫偲側傞丅
- 姵幰偺曋偺宍忬丆攔曋夞悢丆怘帠偺忬懺側偳傪偒傔嵶偐偔僠僃僢僋偟側偑傜丆屄恖偵偁偭偨壓嵻偺搳梌傪峴偆丅
- 壓嵻偲偟偰丆曋傪擃傜偐偔偡傞怹摟埑惈壓嵻乮巁壔儅僌僱僔僂儉丆儔僋僣儘乕僗乯丆挵寰摦塣摦傪懀恑偝偣傞戝挵巋寖惈壓嵻乮僺僐僗儖僼傽乕僩丆僙儞僲僔僪乯偑桳岠偱偁傞丅
- 儖價僾儘僗僩儞偼丆彫挵忋旂嵶朎偵懚嵼偡傞Cl-僠儍僱儖傪妶惈壔偡傞偙偲偵傛傝丆挵娗撪傊偺悈暘暘斿傪懀恑偟偰曋傪擃傜偐偔偟丆挵娗撪桝憲傪崅傔偰攔曋傪懀恑偡傞丅杮朚偱偺揔墳偼枬惈曋旈徢乮婍幙揑幘姵偵傛傞曋旈徢傪彍偔乯偱偁傞偑丆暷崙偱偼枬惈旕偑傫醬捝姵幰偺僆僺僆僀僪桿敪惈挵婡擻忈奞偺帯椕栻偲偟偰彸擣偝傟偰偄傞丅
- 徢忬夵慞偵偼丆壜擻側傜悈暘愛庢丆塣摦丆怘暔慇堐偺愛庢傕桳梡偱偁傞丅
- 忬懺偵墳偠偰煰挵傗揈曋側偳傕峴偆丅
- 僆僺僆僀僪惢嵻傪儌儖僸僱傗僆僉僔僐僪儞偐傜僼僃儞僞僯儖惢嵻偵曄峏偡傞偙偲偱寉夣偡傞偙偲偑偁傞丅
| 暘丂椶 | 栻嵻柤 | 1 擔梡朄丒梡検 | |
|---|---|---|---|
怹摟埑惈壓嵻 |
墫椶壓嵻 | 巁壔儅僌僱僔僂儉 | 1,000乣2,000 mg乮暘2乣3乯 |
| 儔僋僣儘乕僗 | 10乣60 mL乮暘2乣3乯 | ||
| 摐椶壓嵻 | |||
戝挵巋寖惈壓嵻 |
僙儞僫 | 1乣3 g乮暘2乣3乯 | |
| 僙儞僲僔僪 | 12乣48 mg 乮廇怮慜傑偨偼婲彴帪偲廇怮慜乯 |
||
| 僺僐僗儖僼傽乕僩僫僩儕僂儉 | 5乣30 揌/2乣6 忶乮暘2乣3乯 | ||
| 價僒僐僕儖 | 10 mg/夞丆1 擔1乣2 夞乮撢梡乯 | ||
煰挵 |
僌儕僙儕儞 | 10乣150 mL/夞 | |
Cl-僠儍僱儖 |
儖價僾儘僗僩儞 | 48兪g乮暘2乯 | |
乮彫媨岾巕丆壛夑扟敚乯
亂嶲峫暥專亃
1乯 Ishihara M, Ikesue H, Matsunaga H, et al. A multi-institutional study analyzing effect of prophylactic medication for prevention of opioid-induced gastrointestinal dysfunction. Clin J Pain 2012丟28丗373-81
俋丏僆僺僆僀僪偵傛傞暃嶌梡偲懳嶔-偦偺懠偺暃嶌梡偲懳嶔
❶ 柊丂婥
- 僆僺僆僀僪偵傛傞柊婥偼搳梌奐巒弶婜傗憹検帪偵弌尰偡傞偙偲偑懡偄偑丆懴惈偑懍傗偐偵惗偠丆悢擔埲撪偵帺慠偵寉尭側偄偟徚幐偡傞偙偲偑懡偄丅
- 憡屳嶌梡傪娷傓懠偺栻暔丆姶愼徢丆娞丒恡婡擻忈奞丆拞悤恄宱宯偺昦曄丆崅僇儖僔僂儉寣徢側偳丆懠偺尨場傪彍奜偡傞昁梫偑偁傞丅
- 儌儖僸僱偺応崌偼恡婡擻掅壓偵傛傞M6G 偺拁愊偑尨場偲側傞偙偲偑偁傞丅
乵懳丂嶔乶
- 捝傒偑側偔嫮搙偺柊婥偑偁傞応崌偼丆僆僺僆僀僪傪尭検偡傞丅柊婥偺偨傔偵僆僺僆僀僪偺憹検偑崲擄側応崌偼丆僆僺僆僀僪僗僀僢僠儞僌傪専摙偡傞丅
❷ 偣傫栂丒尪妎
- 偑傫姵幰偵偍偄偰偼丆偝傑偞傑側梫場偱偣傫栂仏1 側偳偺擣抦婡擻忈奞偑弌尰偡傞偲偄傢傟偰偍傝丆尨場傪娪暿偡傞昁梫偑偁傞丅
- 僆僺僆僀僪偵傛傞尪妎丆偣傫栂偼搳梌奐巒弶婜傗憹検帪偵弌尰偡傞偙偲偑懡偄丅
- 僆僺僆僀僪埲奜偺尨場栻嵻偲偟偰儀儞僝僕傾僛僺儞宯峈晄埨栻丆峈僐儕儞栻仏2 側偳偵偼摿偵拲堄偑昁梫偱偁傞丅
- 僆僺僆僀僪傪娷傓栻嵻惈偺偣傫栂偼丆尨場栻嵻偺搳梌拞巭偵傛傝悢擔偐傜1 廡娫偱夵慞偡傞応崌偑懡偄丅
- 旕栻嵻惈偺梫場偲偟偰丆揹夝幙堎忢丆拞悤恄宱宯偺昦曄丆姶愼徢丆娞丒恡婡擻忈奞丆掅巁慺徢側偳偑娭梌偟偰偄傞偙偲偑偁傞丅
乵懳丂嶔乶
- 僆僺僆僀僪偑尨場栻嵻偱偁傞壜擻惈偑媈傢傟傞応崌偼丆僆僺僆僀僪偺尭検傗僆僺僆僀僪僗僀僢僠儞僌傪専摙偡傞丅
- 栻暔椕朄偲偟偰僽僠儘僼僃僲儞宯峈惛恄昦栻仏3乮僴儘儁儕僪乕儖側偳乯丆旕掕宆峈惛恄昦栻乮僋僄僠傾僺儞丆僆儔儞僓僺儞側偳乯偺搳梌傪専摙偡傞丅
- 偣傫栂傪惗偠偰偄傞姵幰偑埨怱偱偒傞娐嫬偺挷惍傪峴偆丅
仏1丗偣傫栂
廃埻傪擣幆偡傞堄幆偺惔柧搙偑掅壓偟丆婰壇椡丆尒摉幆忈奞丆尵岅擻椡偺忈奞側偳偺擣抦婡擻忈奞偑婲偙傞忬懺丅捠忢丆悢帪娫偐傜悢擔偺抁婜娫偵敪尰偟丆擔撪曄摦偑戝偒偄丅
仏2丗峈僐儕儞栻
傾僙僠儖僐儕儞偑傾僙僠儖僐儕儞庴梕懱偵寢崌偡傞偺傪慾奞偡傞栻嵻偱丆暃岎姶恄宱傪梷惂偡傞丅嶌梡偑嫮偄栻嵻偱偼偣傫栂傗尪妎側偳偑尰傟傗偡偄丅
仏3丗僽僠儘僼僃僲儞宯峈惛恄昦栻
嫮椡側僪僷儈儞D2庴梕懱慾奞嶌梡傪傕偮丆掕宆峈惛恄昦栻偺堦宯楍丅戙昞揑側栻嵻偲偟偰僴儘儁儕僪乕儖側偳丅
❸ 屇媧梷惂
- 僆僺僆僀僪偵傛傞屇媧梷惂偼丆梡検埶懚揑側墑悜偺屇媧拞悤傊偺捈愙偺嶌梡偵傛傞傕偺偱丆擇巁壔扽慺偵懳偡傞屇媧拞悤偺斀墳偑掅壓偟丆屇媧夞悢偺尭彮偑擣傔傜傟傞丅
- 堦斒揑偵偼偑傫醬捝偺帯椕傪栚揑偲偟偰僆僺僆僀僪傪揔愗偵搳梌偡傞尷傝丆屇媧悢偼掅壓偟側偄偐丆傑偨偼屇媧悢偑掅壓偟偰傕1 夞姺婥検偑憹壛偡傞偺偱掅巁慺寣徢偵側傞偙偲偼傑傟偱偁傞丅偨偩偟丆媫懍惷拲側偳偺搳梌朄偱寣拞擹搙傪媫寖偵忋徃偝偣偨応崌傗醬捝帯椕偵昁梫側検傪戝偒偔忋夞傞夁検搳梌傪峴偭偨応崌偵偼婲偙傝偆傞暃嶌梡偱偁傞丅偟偨偑偭偰丆夁検搳梌偲側傜側偄傛偆偵丆岠壥偲暃嶌梡傪妋擣偟側偑傜憹検傪峴偆昁梫偑偁傞丅傑偨儌儖僸僱搳梌拞丆媫寖偵恡婡擻偑掅壓偡傞偲丆M6G 偺拁愊偵傛傝屇媧梷惂傪惗偠傞壜擻惈偑偁傞丅
- 捝傒偦偺傕偺偑僆僺僆僀僪偺屇媧梷惂偲漢峈偡傞偲偝傟偰偍傝丆奜壢帯椕傗恄宱僽儘僢僋側偳偵傛傝捝傒偑戝暆偵尭彮偁傞偄偼徚幐偟偨応崌偵偼丆憡懳揑偵僆僺僆僀僪偺夁検搳梌偺忬懺偑惗偠丆屇媧梷惂偑敪尰偡傞応崌偑偁傞丅
- 屇媧梷惂偑惗偠傞慜偵偼柊婥傪惗偠傞偨傔丆柊婥傪娤嶡偟丆柊婥偑惗偠偨抜奒偱捔捝庤抜偺尒捈偟偲昡壙傪峴偆偙偲偑廳梫偱偁傞丅
- 僆僺僆僀僪偼丆廳撃側屇媧梷惂偺偁傞姵幰傗丆婥娗巟歜懅敪嶌拞偺姵幰傊偺搳梌偵偮偄偰丆惢嵻偵傛偭偰嬛婖偐怲廳搳梌偲側偭偰偄傞丅
乵懳丂嶔乶
- 巁慺搳梌丆姵幰偺妎惲偲屇媧傪懀偡丅
- 廳撃側応崌偵偼丆栻暔椕朄偲偟偰僆僺僆僀僪漢峈栻偱偁傞僫儘僉僜儞傪巊梡偡傞丅僫儘僉僜儞偼僆僺僆僀僪偵斾傋敿尭婜偑抁偔丆嶌梡帩懕帪娫偼栺30 暘偱偁傞丅偦偺偨傔丆徢忬偺嵞擱偵偁傢偣偰30乣60 暘枅偵暋悢夞搳梌偡傞昁梫偑偁傞丅僫儘僉僜儞偵傛傝捝傒偺埆壔丆嫽暠丆偣傫栂傪惗偠傞偙偲偑偁傞偨傔丆彮検偢偮乮1 夞検偲偟偰0.04乣0.08 mg乯巊梡偡傞丅
❹ 岥撪姡憞
- 僆僺僆僀僪偼丆梡検埶懚揑偵奜暘斿態偵偍偗傞暘斿傪梷惂偡傞丅
- 恑峴偑傫姵幰偺岥撪姡憞偺敪惗昿搙偼30乣97亾偲偝傟傞丅偦偺攚宨偲偟偰丆①懥塼暘斿偺尭彮乮摢栩晹傊偺曻幩慄徠幩丆嶰娐宯峈偆偮栻丆峈僐儕儞栻側偳乯丆②岥峯擲枌偺忈奞乮壔妛椕朄傗曻幩慄帯椕偵傛傞岥撪墛丆岥峯僇儞僕僟徢乯丆③扙悈側偳偑峫偊傜傟傞丅
乵懳丂嶔乶
- 壜擻偱偁傟偽搳梌検偺尭検丆岥撪姡憞傪惗偠傞栻暔偺曄峏傪峴偆丅
- 昿夞偵悈暘傗昘傪愛庢偡傞丆晹壆傪壛幖偡傞側偳悈暘偲幖搙偺曗媼傪峴偄丆恖岺懥塼傗岥峯撪曐幖嵻傪巊梡偡傞丅
- 懥塼暘斿擻偑巆偭偰偄傞応崌丆僉僔儕僩乕儖僈儉傪姎傓側偳丆懥塼態偺暘斿懀恑傪帋傒傞丅
❺ 瘙醳姶
- 僆僺僆僀僪偺峝枌奜搳梌傗僋儌枌壓搳梌偱偼丆懠偺搳梌宱楬偵斾偟偰瘙醳姶偑崅棪偵擣傔傜傟傞丅偙偺斀墳偱偼愐悜屻妏偺僆僺僆僀僪庴梕懱傪夘偟偨婡彉偑峫偊傜傟偰偄傞丅
乵懳丂嶔乶
- 戞堦悽戙偺峈僸僗僞儈儞栻偺搳梌偑堦斒揑偵峴傢傟偰偄傞偑丆柍岠偱偁傞偙偲偑懡偄丅
- 僆儞僟儞僙僩儘儞側偳5-HT3庴梕懱漢峈栻偑桳岠側応崌偑偁傞丅
- 奜梡嵻偲偟偰偼垷墧壺擃峱丆僒儕僠儖巁擃峱傗0.25乣2亾偺儊儞僩乕儖偺崿崌惢嵻偑桳梡偲偝傟偰偄傞丅
- 嶤夁偵傛傞旂晢忈奞偑嫮偄応崌偼丆庛乣拞摍搙偺僐儖僠僐僗僥儘僀僪奜梡嵻偺巊梡傕峫椂偡傞丅嫮僐儖僠僐僗僥儘僀僪奜梡嵻偺挿婜搳梌偼丆旂晢偺堔弅傗擇師姶愼傪惗偠傞偙偲偑偁傞偨傔丆抁婜偺巊梡偵偲偳傔傞傋偒偱偁傞丅
- 擇師揑側姶愼傪嵟彫尷偵偡傞偨傔偵丆捾傪抁偔愗偭偨庤偱寉偔偙偡傞丆庤戃傪拝梡偡傞側偳丆擔忢峴摦偺嫵堢傕廳梫偱偁傞丅
- 徢忬偺夵慞偑傒傜傟側偄応崌偼僆僺僆僀僪僗僀僢僠儞僌傪専摙偡傞丅
❻ 攔擜忈奞
- 僆僺僆僀僪偺搳梌偵傛傝擜娗偺嬞挘傗廂弅傪憹壛偝偣傞偙偲偑偁傞丅
- 僆僺僆僀僪偼攔擜斀幩傪梷惂偟丆奜擜摴妵栺嬝偺廂弅偍傛傃銷泖梕検傪偲傕偵憹壛偝偣傞丅
- 攔擜忈奞偼崅楊偺抝惈偵懡偔擣傔傜傟丆慜棫態旍戝徢偺姵幰偱偼擜暵偵帄傞応崌傕偁傝拲堄偑昁梫偱偁傞丅
乵懳丂嶔乶
- 栻暔椕朄偲偟偰攔擜嬝偺廂弅傪崅傔傞僐儕儞嶌摦栻傗丆妵栺嬝傪抩娚偝偣傞兛1 庴梕懱幷抐栻仏1 偺搳梌偑峴傢傟傞偙偲偑偁傞丅
仏1丗兛1庴梕懱幷抐栻
3 偮偵暘椶偝傟傞傾僪儗僫儕儞庴梕懱乮兛1丆兛2丆兝乯偺偆偪丆兛1庴梕懱偺傒偵幷抐嶌梡傪帵偡栻嵻丅兛1庴梕懱偼庡偵寣娗丒擜楬側偳偺暯妸嬝偵懚嵼偡傞丅崅寣埑丒攔擜忈奞側偳偑庡側揔墳徢偱偁傞丅
❼ 儈僆僋儘乕僰僗
- 僆僺僆僀僪搳梌帪偵儈僆僋儘乕僰僗仏2 偑敪尰偡傞偙偲偑偁傞丅
- 儈僆僋儘乕僰僗偲偼丆1 偮偁傞偄偼暋悢偺嬝擏偑抁帪娫偱偁傞偑晄悘堄偵廂弅偡傞傕偺偱偁傞乮巐巿偑僺僋僢偲偡傞側偳乯丅
- 儌儖僸僱偺応崌丆恄宱撆惈偺偁傞戙幱暔偺拁愊偑梫場偺堦偮偲峫偊傜傟偰偄傞丅
乵懳丂嶔乶
- 栻暔椕朄偲偟偰偼僋儘僫僛僷儉丆儈僟僝儔儉側偳偑桳岠側応崌偑偁傞丅
- 僆僺僆僀僪僗僀僢僠儞僌傪専摙偡傞丅
仏2丗儈僆僋儘乕僰僗
晄悘堄塣摦偺堦庬丅1 偮偁傞偄偼暋悢偺嬝擏偑摨帪偵慺憗偔廂弅偡傞丅慡恎偁傞偄偼摿掕偺晹埵偵偩偗偵婲偙傞応崌偑偁傞丅
❽ 捝妎夁晀乮hyperalgesia乯
- 捝妎夁晀乮hyperalgesia乯偲偼丆捠忢捝傒傪姶偠傞巋寖偵傛偭偰桿敪偝傟傞斀墳偑丆捠忢傛傝傕嫮偔側偭偨忬懺偺偙偲傪偄偆乮Ⅱ-1-1-3 恄宱忈奞惈醬捝偺崁嶲徠乯丅
- 戝検偺僆僺僆僀僪傪峝枌奜搳梌偡傞偙偲偵傛傝丆傑傟偵惗偠傞偙偲偑偁傞偲偄傢傟偰偄傞丅
- 尨場偲偟偰丆僆僺僆僀僪戙幱暔側偳偺娭梌偑峫偊傜傟偰偄傞丅
- 僆僺僆僀僪偑尨場偺捝妎夁晀偺忬懺偱偼僆僺僆僀僪傪憹検偡傞偲捝傒偑埆壔偡傞丅
- 僆僺僆僀僪偺憹検偵敽偄媫寖偵捝傒偑憹嫮偡傞応崌偼捝妎夁晀偺壜擻惈傪峫椂偡傞丅
乵懳丂嶔乶
- 僆僺僆僀僪偺尭検傑偨偼拞巭丆僆僺僆僀僪埲奜偺捔捝栻丆僆僺僆僀僪僗僀僢僠儞僌傪専摙偡傞丅
❾ 怱寣娗宯偺暃嶌梡
- 儊僒僪儞傪巊梡偡傞偙偲偵傛傝丆QT 墑挿傗怱幒昿攺乮Torsades de pointes 傪娷傓乯偑敪尰偡傞偙偲偑偁傞丅
乵懳丂嶔乶
- 儊僒僪儞傪搳梌奐巒慜丒搳梌拞偼丆掕婜揑偵怱揹恾専嵏偍傛傃揹夝幙専嵏傪峴偆丅
- 摿偵丆儊僒僪儞偺1 擔搳梌検偑100 mg 傪挻偊傞慜偍傛傃偦偺1 廡娫屻丆QT 墑挿傪婲偙偟傗偡偄姵幰偱偼丆儊僒僪儞偺搳梌検偑埨掕偟偨帪揰偱怱揹恾専嵏傪峴偆偙偲偑朷傑偟偄丅
乮壀杮掯峎乯
10丏僆僺僆僀僪偵梌偊傞塭嬁丒栻暔憡屳嶌梡
❶ 栻暔憡屳嶌梡偲偼
栻暔憡屳嶌梡乮埲壓丆憡屳嶌梡乯偲偼丆偁傞庬偺栻暔偺岠壥偑懠偺栻暔傪暪梡偡傞偙偲偵傛傝戝偒偔曄壔偡傞偙偲傪偄偆丅偡側傢偪丆2 庬椶埲忋偺栻暔傪暪梡偡傞偙偲偱丆栻暔偺岠壥偑撆惈椞堟偵傑偱憹嫮偡傞偙偲傗丆偦偺斀懳偵栻暔偵傛傞帯椕岠壥偑尭庛偡傞偙偲傪偄偆丅
偙偺偨傔丆栻暔搳梌偵敽偄梊憐奜偺斀墳偑弌尰偟偨応崌偼丆忢偵憡屳嶌梡傪媈偆昁梫偑偁傞丅憡屳嶌梡偼丆栻暔摦懺妛揑憡屳嶌梡偲栻椡妛揑憡屳嶌梡偺2 庬椶偵戝暿偱偒丆偙傟傜傪婎偵偦偺婡彉傪棟夝偡傞偙偲偱丆偁傜偐偠傔敪尰傪梊應偡傞偙偲偑壜擻偲側傞丅
乵栻暔摦懺妛揑憡屳嶌梡pharmacokinetic drug interaction乶丂栻暔A 偑栻暔B 偺媧廂丆暘晍丆戙幱丆攔煏偵塭嬁傪梌偊傞寢壥丆嶌梡晹埵偱偺栻暔B 偺擹搙偑曄壔偟丆偦偺岠壥偑憹嫮傑偨偼尭庛偡傞傛偆側応崌傪偄偆丅
乵栻椡妛揑憡屳嶌梡pharmacodynamic drug interaction乶丂栻暔A 偲栻暔B 偑嶌梡晹埵偱嫤椡偁傞偄偼漢峈偡傞応崌傪偄偆丅嫤椡嶌梡偵偼憡壛嶌梡乮岠壥偑奺栻暔偺岠壥偺榓乯偲憡忔嶌梡乮岠壥偑奺栻暔偺岠壥偺榓埲忋乯偑偁傞丅
❷ 僆僺僆僀僪巊梡帪偵拲堄偡傋偒憡屳嶌梡乮昞8乯
僆僺僆僀僪偼丆拞悤恄宱梷惂栻乮僼僃僲僠傾僕儞桿摫懱丆僶儖價僣乕儖巁桿摫懱丆 儀儞僝僕傾僛僺儞宯栻嵻側偳乯丆媧擖杻悓栻丆MAO 慾奞栻丆嶰娐宯峈偆偮栻丆兝幷抐栻丆傾儖僐乕儖丆峈僸僗僞儈儞栻偲偺暪梡偵傛傝憡壛揑偵拞悤恄宱梷惂嶌梡傪憹嫮偡傞偨傔丆暪梡帪偼屇媧梷惂丆傔傑偄丆掅寣埑偍傛傃捔惷偵拲堄偡傞昁梫偑偁傞丅僆僺僆僀僪偼丆峈僐儕儞嶌梡傪桳偡傞栻暔偲暪梡偡傞偙偲偵傛傝杻醿惈僀儗僂僗偵帄傞廳撃側曋旈傑偨偼擜暵側偳傪婲偙偡壜擻惈偑偁傞丅偦偺懠丆僆僺僆僀僪偼丆杻栻漢峈惈捔捝栻仏偱偁傞僽僾儗僲儖僼傿儞傗儁儞僞僝僔儞偲暪梡偡傞偙偲偱僆僺僆僀僪庴梕懱傊偺寢崌偑慾奞偝傟丆捔捝嶌梡偺尭庛傗棧扙徢岓偑敪尰偡傞壜擻惈偑偁傞丅偦偺偨傔丆尨懃偲偟偰椉幰傪暪梡偡傋偒偱偼側偄丅
仏丗杻栻漢峈惈捔捝栻
僆僺僆僀僪嶌摦栻偑懚嵼偟側偄忬嫷偱偼嶌摦栻偲偟偰嶌梡偡傞偑丆僆僺僆僀僪嶌摦栻偺懚嵼壓偱偼偦偺嶌梡偵漢峈偡傞嶌梡傪傕偮捔捝栻丅
❸ 摿偵儌儖僸僱丒僆僉僔僐僪儞丒僼僃儞僞僯儖丒儊僒僪儞巊梡帪偵拲堄偡傋偒憡屳嶌梡
僉僯僕儞偼丆媧廂夁掱偵偍偗傞栻暔憡屳嶌梡偵傛傝儌儖僸僱宱岥惢嵻偺栻暔寣拞擹搙乮帪娫乯嬋慄壓柺愊乮area under the drug concentration time curve丟AUC乯偲嵟崅寣拞擹搙乮maximum drug concentration丟Cmax乯傪忋徃偝偣傞丅堦曽丆儕僼傽儞僺僔儞偼儌儖僸僱偺嶌梡傪尭庛偝偣傞偙偲偑偁傞丅偦偺偨傔丆偙傟傜偺栻暔偲儌儖僸僱傪暪梡偡傞応崌偼丆儌儖僸僱偺捔捝岠壥傪廫暘偵娤嶡偡傞昁梫偑偁傞丅
僆僉僔僐僪儞偼丆CYP2D6 慾奞栻偲暪梡偟偨応崌O-扙儊僠儖壔斀墳偑慾奞偝傟丆偦偺寢壥僆僉僔僐僪儞偺寣拞擹搙偑崅傑傞壜擻惈偑偁傞丅椺偊偽丆慖戰揑僙儘僩僯儞嵞庢傝崬傒慾奞栻偺懡偔偼CYP2D6 慾奞栻偱偁傞偨傔丆戙幱夁掱偵偍偗傞栻暔摦懺妛揑憡屳嶌梡偵傛傝僆僉僔僐僪儞偺岠壥偑嫮傑傞壜擻惈偑偁傞丅儃儕僐僫僝乕儖側偳偺CYP3A4 慾奞栻傕傑偨丆暪梡偟偨応崌N-扙儊僠儖壔斀墳傪慾奞偡傞偨傔丆僆僉僔僐僪儞偺寣拞擹搙傪忋徃偝偣傞壜擻惈偑偁傞丅傑偨丆儕僼傽儞僺僔儞偼僆僉僔僐僪儞偺僋儕傾儔儞僗傪憹壛偝偣寣拞擹搙傪掅壓偝偣傞丅堦曽丆僆僉僔僐僪儞偦偺傕偺偼僔僋儘僗億儕儞偺惗懱撪棙梡棪傪尭彮偝偣傞偨傔丆暪梡偡傞応崌偵偼僔僋儘僗億儕儞偺栻岠偑尭庛偡傞丅
僼僃儞僞僯儖偼娞栻暔戙幱峺慺CYP3A4 偱戙幱偝傟傞偨傔丆杮峺慺傪慾奞偡傞儕僩僫價儖丆傾儈僆僟儘儞丆僋儔儕僗儘儅僀僔儞丆僕儖僠傾僛儉丆僼儖儃僉僒儈儞丆偝傜偵丆僀僩儔僐僫僝乕儖丆僼儖僐僫僝乕儖丆儃儕僐僫僝乕儖側偳偺僩儕傾僝乕儖宯峈恀嬠栻偲暪梡偡傞偙偲偵傛傝丆栻暔摦懺妛揑側憡屳嶌梡偱僼僃儞僞僯儖偺AUC 偺憹壛傗寣拞擹搙敿尭婜偺墑挿傪堷偒婲偙偡丅
儊僒僪儞偼庡偵CYP3A4 偲CYP2B6 偱戙幱偝傟丆CYP2D6丆CYP2C9丆CYP2C19 側偳偱傕戙幱偝傟傞丅偦偺偨傔丆奺庬栻暔戙幱峺慺偑娭學偡傞慾奞丒桿摫嶌梡偵傛傝丆儊僒僪儞偺寣拞擹搙偑忋徃傑偨偼掅壓偡傞壜擻惈偑偁傞丅儊僒僪儞偲僯儏乕僉僲儘儞宯峈嬠栻偺僔僾儘僼儘僉僒僔儞傗僩儕傾僝乕儖宯峈恀嬠栻傪暪梡偡傞偙偲偵傛傝丆儊僒僪儞偺寣拞擹搙偑忋徃偡傞壜擻惈偑偁傞丅僒僉僫價儖傗僄僼傽價儗儞僣側偳偺峈HIV 栻偲偺暪梡偱偼丆儊僒僪儞偺寣拞擹搙偑掅壓偡傞偲偺曬崘偑偁傞丅傑偨丆暃嶌梡偲偟偰QT 墑挿傗Torsade de pointes 偺偁傞栻嵻偲儊僒僪儞偲偺暪梡偼丆怱撆惈偺暃嶌梡敪尰偺儕僗僋傪崅傔傞丅
庡側僆僺僆僀僪 暪梡栻 |
儌儖僸僱 | 僆僉僔 僐僪儞 |
僼僃儞僞 僯儖 |
儊僒僪儞 | 庡側婡彉 |
|---|---|---|---|---|---|
拞悤恄宱梷惂栻 |
↑ | ↑ | ↑ | ↑ | 拞悤梷惂嶌梡 |
峈嬅屌栻 |
↑ | ↑ | 晄柧 |
||
杻栻漢峈惈捔捝栻 |
仱 | 仱 | 仱 | 庴梕懱寢崌偺 |
|
CYP2D6 慾奞栻 |
仮 | 仮 | 娞戙幱偺曄壔 |
||
CYP3A4 慾奞栻 |
仮 | 仮 | 仮 | 娞戙幱偺曄壔 |
↑/↓丗暪梡栻偺嶌梡憹嫮/尭庛
仮/仱丗僆僺僆僀僪偺嶌梡憹嫮/尭庛
11丏旕僗僥儘僀僪惈徚墛捔捝栻巊梡帪偵拲堄偡傋偒憡屳嶌梡乮昞9乯
旕僗僥儘僀僪惈徚墛捔捝栻乮NSAIDs乯偺懡偔偼丆寣塼拞偱偼戝晹暘偑寣燋抈敀偲寢崌偟偨忬懺偱懚嵼偡傞丅偟偨偑偭偰丆抈敀寢崌惈偑崅偄栻暔偑摨帪偵搳梌偝傟偨応崌丆抈敀寢崌偺嫞崌偑婲偙傝丆寣燋抈敀偲寢崌偟偰偄側偄梀棧宆偺栻暔偺妱崌偑憹壛偟丆偦偺栻暔偺嶌梡偑憹嫮偡傞壜擻惈偑偁傞丅
NSAIDs 偼庡偵娞憻偵偍偄偰戙幱偝傟傞偨傔丆摨堦峺慺偵傛偭偰戙幱偝傟傞栻暔偑暪梡偝傟偨応崌丆戙幱夁掱偵偍偗傞栻暔摦懺妛揑憡屳嶌梡偵傛傝丆峺慺偵懳偟偰嫞崌揑寢崌偑惗偠傞丅偦偺寢壥丆栻暔偺寣拞擹搙偑崅傑傝丆嶌梡偑嫮偔尰傟傞応崌偑偁傞丅椺偊偽丆僙儗僐僉僔僽偼CYP2C9 偱戙幱偝傟傞偨傔丆摨峺慺偱戙幱偝傟傞儚儖僼傽儕儞偺峈嬅屌嶌梡傪憹嫮偟丆摿偵崅楊幰偱偼弌寣孹岦傪崅傔傞壜擻惈偑偁傞丅懡偔偺NSAIDs 偼儚儖僼傽儕儞傗懠偺僋儅儕儞偺峈嬅屌嶌梡傪帪偵憹嫮偟丆廳搙偺弌寣傪敪尰偡傞偙偲偑曬崘偝傟偰偄傞丅偦偺偨傔丆NSAIDs 偲儚儖僼傽儕儞傪暪梡偡傞応崌偼拲堄怺偔嬅屌擻傪儌僯僞儕儞僌偡傞昁梫偑偁傞丅摨條偵堦晹偺NSAIDs 偼丆僼僃僯僩僀儞傗僗儖儂僯儖擜慺宯寣摐崀壓栻乮SU 栻乯偲暪梡偟偨応崌丆偦傟傜偺嶌梡傪憹嫮偡傞壜擻惈偑偁傞偙偲偑抦傜傟偰偄傞丅傑偨NSAIDs 偲偺擜嵶娗暘斿偺嫞崌偵傛傝丆儊僩僩儗僉僒乕僩丆儕僠僂儉丆僕僊僞儕僗偺攔煏抶墑偑惗偠丆偦傟傜偺嶌梡傪憹嫮偡傞丅NSAIDs 偼寣娗奼挘嶌梡傗僫僩儕僂儉棙擜嶌梡傪桳偡傞僾儘僗僞僌儔儞僕儞偺崌惉傪梷惂偡傞偙偲偐傜丆ACE 慾奞栻乮傾儞僕僆僥儞僔儞曄姺峺慺慾奞栻乯傗棙擜栻偺岠壥傪尭庛偝偣傞丅摿偵丆ACE 慾奞栻偺暪梡偱偼丆恡婡擻忈奞偺儕僗僋傪忋徃偝偣丆傑傟偵崅僇儕僂儉寣徢傪棃偡偙偲偑偁傞丅
NSAIDs 偲僯儏乕僉僲儘儞宯峈嬠栻仏1 傪暪梡偡傞偲丆僯儏乕僉僲儘儞宯峈嬠栻偺拞悤恄宱嶌梡偱偁傞兞-傾儈僲棌巁庴梕懱乮GABA 庴梕懱乯墳摎梷惂嶌梡偵傛傝鑷抣傪掅壓偝偣丆偗偄傟傫傪桿敪偡傞偙偲偑偁傞丅摿偵偰傫偐傫姵幰傗慺場偺偁傞姵幰偱偼偗偄傟傫偺儕僗僋偑憹壛偡傞壜擻惈偑偁傞丅NSAIDs 偲儁儊僩儗僉僙僪偲偺暪梡偼儁儊僩儗僉僙僪偺恡攔煏傪掅壓偝偣傞壜擻惈偑偁傞偺偱拲堄偡傞丅堦曽丆儈僜僾儘僗僩乕儖偼丆媧廂慾奞傪婲偙偡偙偲偵傛傝丆僕僋儘僼僃僫僋僫僩儕僂儉偺AUC 偲Cmax 傪壓崀偝偣傞丅峈寣彫斅椕朄偵敽偆掅梡検傾僗僺儕儞偲NSAIDs丆椺偊偽僀僽僾儘僼僃儞偲偺暪梡偱偼丆僀僽僾儘僼僃儞偵傛偭偰寣彫斅偺COX-1 偺妶惈晹埵偑愭偵愯桳偝傟傞偲丆傾僗僺儕儞偑寣彫斅偺昗揑晹埵偵寢崌偱偒側偄偨傔晄壜媡揑側寣彫斅婡擻慾奞偑婲偙傜側偔側傝丆傾僗僺儕儞偺峈寣彫斅嶌梡偑敪婗偝傟側偔側傞壜擻惈偑偁傞丅傑偨丆2 庬椶埲忋偺NSAIDs 傪暪梡搳梌偡傞偙偲偱徚壔娗忈奞偺儕僗僋偑憹壛偡傞偙偲偵傕拲堄偡傞丅
仏1丗僯儏乕僉僲儘儞宯峈嬠栻
恖岺崌惉偝傟偨峈嬠栻偺堦宯楍丅嵶嬠偺DNA 暋惢偵昁恵偺峺慺乮DNA 僕儍僀儗乕僗側偳乯傪慾奞偟嶦嬠揑偵嶌梡偡傞丅暆峀偄峈嬠僗儁僋僩儖偲嫮偄峈嬠椡偑摿挜乮戙昞揑側栻嵻偲偟偰僆僼儘僉僒僔儞丆儗儃僼儘僉僒僔儞側偳乯丅
庡側NSAIDs 暪梡栻 |
僙儗僐僉僔僽 |
儊儘僉僔僇儉 |
儘僉僜僾儘僼僃儞 |
僀僽僾儘僼僃儞 |
僼儖儖價僾儘僼僃儞 |
僕僋儘僼僃僫僋 |
庡側婡彉 |
|---|---|---|---|---|---|---|---|
儚儖僼傽儕儞 |
↑ | ↑ | ↑ | ↑ | ↑ | ↑ | 娞戙幱偺曄壔 |
儊僩僩儗僉僒乕僩 |
↑ | ↑ | ↑ | 恡攔煏偺曄壔 |
|||
ACE 慾奞栻 |
↓ | ↓ | 恡偵偍偗傞僾儘僗僞僌儔儞僕儞崌惉慾奞 |
||||
僒僀傾僓僀僪宯棙擜栻 |
↓ | ↓ | ↓ | ↓ | ↓ | ↓ | 恡偵偍偗傞僾儘僗僞僌儔儞僕儞崌惉慾奞 |
儖乕僾棙擜栻 |
↓ | ↓ | ↓ | ↓ | ↓ | ↓ | 恡偵偍偗傞僾儘僗僞僌儔儞僕儞崌惉慾奞 |
僕僑僉僔儞 |
↑ | 恡攔煏偺曄壔 |
|||||
僗儖儂僯儖擜慺栻乮SU栻乯 |
仮 | ↑ | 恡攔煏偺曄壔 |
||||
僯儏乕僉僲儘儞宯峈嬠栻 |
× | 庴梕懱寢崌偺曄壔 |
|||||
儁儊僩儗僉僙僪 |
↑ | ↑ | ↑ | ↑ | ↑ | ↑ | 恡攔煏偺曄壔 |
儈僜僾儘僗僩乕儖 |
仱 | 媧廂偺曄壔 |
↑/↓丗暪梡栻偺嶌梡憹嫮/尭庛
仮/仱丗NSAIDs 偺嶌梡憹嫮
×丗 儘儊僼儘僉僒僔儞丆僲儖僼儘僉僒僔儞丆僾儖儕僼儘僉僒僔儞偺傒暪梡嬛婖丆懠偼暪梡拲堄
乲奺栻嵻偺揧晅暥彂傛傝嶌惉乴
12丏僆僺僆僀僪偲怘帠偺塭嬁
僆僺僆僀僪偼堦斒揑偵怘帠偺帪娫偵娭學側偔丆掕婜揑偵暈梡偡傞偙偲偑悇彠偝傟 偰偄傞丅堦晹偺僆僺僆僀僪惢嵻偼丆怘帠乮崅帀朾怘仏2 愛庢乯偑媧廂偵塭嬁乮栻暔摦懺妛揑憡屳嶌梡乯傪媦傏偡壜擻惈偑偁傞偲曬崘偝傟偰偄傞丅僺乕僈乕僪® 偼丆崅帀朾怘愛庢20 暘屻傑偨偼寉怘愛庢30 暘慜搳梌偱偼丆嬻暊帪搳梌偲斾傋偰AUC 偍傛傃Cmax偑栺50亾掅壓丆僷僔乕僼® 偼崅帀朾怘愛庢屻偱偼AUC 偑栺18亾尭彮丆僇僨傿傾儞® 偼怘屻偵暈梡偡傞偲丆嬻暊帪偵暈梡偟偨帪偲斾妑偟偰嵟崅寣拞擹搙摓払帪娫乮maximum drug concentration time丟Tmax乯偑栺1.6 帪娫抶墑偡傞偙偲傕偁傞丅偦偺懠偵僆僉僔僐僪儞懍曻惈惢嵻乮僆僉僲乕儉®乯偼丆崅帀朾怘愛庢屻偵AUC 偑栺20亾忋徃偡傞丅怘帠偵傛傞塭嬁傗惢嵻偺摿挜傪棟夝偟偨偆偊偱丆姵幰偺徢忬傗惗妶僗僞僀儖偵偁傢偣偨栻暔搳梌愝寁偑昁梫偱偁傞丅
傑偨丆僒僾儕儊儞僩偲偟偰愛庢偝傟傞僙僀儓僂僆僩僊儕僜僂乮僙儞僩丒僕儑乕儞僘丒儚乕僩乯偼丆儊僒僪儞偺寣拞擹搙傪掅壓偝偣偰棧扙徢忬傪弌尰偝偣偨偲偺曬崘偑偁傞丅傾儖僐乕儖偼丆僆僺僆僀僪偺拞悤恄宱梷惂嶌梡傪憹嫮偡傞偨傔丆柊婥偺憹嫮傗妎惲搙偺掅壓丆応崌偵傛偭偰偼廳撃側屇媧梷惂傪堷偒婲偙偡偙偲偐傜丆廫暘側拲堄偑昁梫偱偁傞丅
乮钹悾媣岝丆嵶扟丂帯丆愒栘丂揙丆嵅栰尦旻乯
仏2丗崅帀朾怘
帀朾偑懡偔娷傑傟傞怘昳傪偲傞怘帠丅柧妋側掕媊偼側偄偑丆憤愛庢僄僱儖僊乕偺偆偪帀朾偑愯傔傞妱崌乮帀朾僄僱儖僊乕斾棪乯偑偍偍傛偦30乣40亾埲忋偲偝傟傞偙偲偑懡偄丅
13丏惛恄埶懚丒恎懱埶懚丒懴惈
僆僺僆僀僪偵娭偡傞岆夝偑醬捝帯椕偺忈奞偲側偭偰偍傝丆惛恄埶懚乮psychological dependence乯丆恎懱埶懚乮physical dependence乯丆懴惈乮tolerance乯偺3 偮偺奣擮傪惓偟偔棟夝偡傞偙偲偑廳梫偱偁傞丅
❶ 掕丂媊
惛恄埶懚丆恎懱埶懚丆懴惈偵娭偡傞掕媊偼崙嵺揑偵傕摑堦偝傟偰偄側偄丅杮僈僀僪儔僀儞偱偼丆American Pain Society丆American Academy of Pain Medicine丆American Society of Addiction Medicine 偑摑堦偟偨尒夝傪傑偲傔傞偨傔偵愝抲偟偨Liaison Committee on Pain and Addiction 偺姪崘傪嶲峫偵丆愱栧壠偺崌堄偵婎偯偒丆埲壓偺掕媊傪梡偄傞丅
1 乯惛恄埶懚
師偺偆偪偄偢傟偐1 偮傪娷傓峴摦偵傛偭偰摿挜偯偗傜傟傞堦師惈偺枬惈恄宱惗暔妛揑幘姵偱偁傞丅偦偺敪尰偲挜岓偵塭嬁傪媦傏偡堚揱揑丆怱棟丒幮夛揑丆娐嫬揑梫慺偑偁傞丅
乮Savage 傜偺掕媊乯- ①帺屓惂屼偱偒偢偵栻暔傪巊梡偡傞
- ②徢忬乮捝傒乯偑側偄偵傕偐偐傢傜偢嫮敆揑偵栻暔傪巊梡偡傞
- ③桳奞側塭嬁偑偁傞偵傕偐偐傢傜偢帩懕偟偰巊梡偡傞
- ④栻暔偵懳偡傞嫮搙偺梸媮偑偁傞
乵夝丂愢乶丂偙傟傜偼丆Savage 傜偵傛傝嶌惉偝傟偨乽歯暼乮addiction乯乿偺掕媊偱偁傞丅擔杮偱偼丆乽杻栻媦傃岦惛恄栻庢掲朄戞2 忦擇廫巐丆擇廫屲乿偵偍偄偰丆乽杻栻拞撆丗杻栻丆戝杻枖偼偁傊傫偺枬惈拞撆傪偄偆丅乿丆乽杻栻拞撆幰丗杻栻拞撆偺忬懺偵偁傞幰傪偄偆丅乿偲弎傋傜傟偰偄傞丅偙偙偱偺乽拞撆乮杻栻拞撆乯乿偲偄偆梡岅偼朄棩忋偺梡岅偱偁傞丅堛妛揑偵乽拞撆乿偲偼埶懚惈偲偼娭學側偔丆戝検搳梌帪偁傞偄偼枬惈揑偵搳梌偟偨帪偵尰傟傞桳奞帠徾偱偁傝丆乽杻栻偍傛傃岦惛恄栻庢掲朄乿偱弎傋傜傟偰偄傞乽杻栻拞撆乿偼丆乽歯暼乮addiction乯乿偵嬤偄奣擮偱偁傞偲峫偊傜傟傞丅傑偨丆乽偑傫娚榓働傾僈僀僪僽僢僋乿乮擔杮堛巘夛娔廋乯偱偼乽拞撆乮杻栻拞撆乯乿傪埲壓偺傛偆偵掕媊偟偰偍傝丆偙傟偼丆Portenoy 傜偺乽addiction乮歯暼乯乿偺掕媊傪朚栿偟偨傕偺偱偁傞丅
埲壓偺傛偆側摿挜傪傕偮怱棟揑丆峴摦揑側徢岓孮偲掕媊偡傞丅
偐偮/傑偨偼 |
堦曽丆WHO 偺摑寁婎弨偵婎偯偒暘椶偝傟偨幘昦傗巰場偺暘椶偱偁傞ICD-10 偱偼乽歯暼乮addiction乯乿偼乽埶懚徢岓孮乮dependence syndrome乯乿偲偄偆梡岅偲偟偰丆傑偨丆惛恄堛妛偺椞堟偵偍偄偰ICD-10 偲側傜傃戙昞揑側恌抐婎弨偺堦偮偱偁傞DSM-Ⅳ偱偼乽暔幙埶懚乮substance dependence乯乿偲偄偆梡岅偱掕媊偝傟偰偄傞丅
| 偁傞暔幙偁傞偄偼偁傞庬偺暔幙巊梡偑丆偦偺恖偵偲偭偰埲慜偵偼傛傝戝偒側壙抣傪傕偭偰偄偨懠偺峴摦傛傝丆偼傞偐偵桪愭偡傞傛偆偵側傞堦孮偺惗棟揑丆峴摦揑丆擣抦揑尰徾偺偙偲偱偁傞丅埶懚徢岓孮偺拞怱偲側傞摿挜偼丆惛恄嶌梡暔幙乮堛妛揑偵張曽偝傟偨傕偺偱偁偭偰傕側偔偰傕乯丆傾儖僐乕儖偁傞偄偼僞僶僐傪巊梡偟偨偄偲偄偆梸朷乮偟偽偟偽嫮偔丆帪偵掞峈偱偒側偄乯偱偁傞丅偁傞婜娫暔幙傪棧扙偟偨偁偲偵嵞巊梡偡傞偲丆旕埶懚幰傛傝傕憗偔偙偺徢岓孮偺懠偺摿挜偑嵞弌尰偡傞偙偲偑柧傜偐偵偝傟偰偄傞丅 |
| 椪彴揑偵廳戝側忈奞傗嬯捝傪堷偒婲偙偡暔幙偺晄揔愗側巊梡偵敽偭偰丆埲壓偺3 偮乮傑 偨偼偦傟埲忋乯偑丆摨偠12 僇寧偺婜娫撪偺偳偙偐偱婲偙傞偙偲偵傛偭偰帵偝傟傞丅
|
拲丗2013 擭5 寧偵DSM-Ⅴ偑敪峴偝傟偨偑丆僊儍儞僽儖傗僀儞僞乕僱僢僩側偳偺暔幙偱偼側偄傕偺偑擖偭偨偨傔偵暔幙埶懚偲偄偆尵梩偑側偔側傝丆Substance Use and Addictive Disorders乮暔幙巊梡偍傛傃傾僨傿僋僔儑儞偺忈奞孮乯偑梡偄傜傟偰偄傞丅
乽拞撆乮杻栻拞撆乯乿偺掕媊偼丆妛夛傗抍懱偵傛偭偰梡岅丆掕媊偑傑偪傑偪偱摑堦偝傟偰偄側偄丅杮僈僀僪儔僀儞偱偼丆偙傟傜偺側偐偱嵟傕娙寜側Savage 傜偺乽歯暼乮addiction乯乿偺掕媊傪丆乽惛恄埶懚乿偲偄偆堦斒揑偵傢偐傝傗偡偔丆偐偮堛妛揑側乽拞撆乿偲嬫暿偱偒傞昞尰傪梡偄偰掕媊偟偨丅傑偨丆乽拞撆乮杻栻拞撆乯乿偲偄偆朄棩梡岅偼堛妛揑側媫惈拞撆傪堄枴偡傞乽拞撆乿偲堎側傝丆棟夝偺崿棎傪惗偠偝偣傞尨場偲側傞偨傔巊梡偟側偄偙偲偲偟偨丅
2 乯恎懱埶懚
乵掕丂媊乶丂撍慠偺栻暔拞巭丆媫懍側搳梌検尭彮丆寣拞擹搙掅壓丆偍傛傃漢峈栻搳梌偵傛傝偦偺栻暔偵摿桳側棧扙徢岓孮偑惗偠傞偙偲偵傛傝柧傜偐偵偝傟傞丆恎懱偺栻暔偵懳偡傞惗棟揑弴墳忬懺偱偁傞丅
乵夝丂愢乶丂恎懱埶懚偼丆僆僺僆僀僪偵尷傜偢挿婜娫栻暔偵敇業偝傟傞偙偲偵傛偭偰惗偠傞惗懱偺惗棟妛揑側揔墳忬懺偱偁傞丅恎懱埶懚偑惗偠偰偄傞偐偳偆偐偼丆栻暔傪拞巭偟偨応崌偵丆栻暔偵摿挜揑側棧扙徢岓孮偑惗偠傞偙偲偱敾抐偡傞丅偡側傢偪丆栻暔傪拞巭偟偨帪偵棧扙徢岓偑傒傜傟傟偽恎懱埶懚偑宍惉偝傟偰偄傞偙偲傪帵偡丅僆僺僆僀僪偺応崌丆壓棢丆旲楻丆敪娋丆恎恔偄傪傆偔傓帺棩恄宱徢忬偲丆拞悤恄宱徢忬偑棧扙徢岓孮偲偟偰婲偙傞丅
恎懱埶懚傪宍惉偡傞栻暔偼僆僺僆僀僪偺傒偱偼側偔丆僶儖價僣乕儖巁丆傾儖僐乕儖偑偁傞丅偝傜偵丆僯僐僠儞傕庛偄恎懱埶懚傪帵偡丅
恎懱埶懚偼僆僺僆僀僪偺挿婜搳梌傪庴偗傞偑傫姵幰偺懡偔偱擣傔傜傟傞偑丆捝傒偺偨傔偵僆僺僆僀僪偑搳梌偝傟偰偄傟偽惗懱偵晄棙塿傪惗偠側偄偙偲丆惛恄埶懚偲偼堎側傞偙偲丆僆僺僆僀僪埲奜偺栻暔偱傕惗偠傞惗棟揑側弴墳忬懺偱偁傞偙偲傪棟夝偡傞昁梫偑偁傞丅
3 乯懴丂惈
乵掕丂媊乶丂弶婜偵搳梌偝傟偰偄偨栻暔偺梡検偱摼傜傟偰偄偨栻棟妛揑岠壥偑帪娫宱夁偲偲傕偵尭戅偟丆摨偠岠壥傪摼傞偨傔偵傛傝懡偔偺梡検偑昁梫偵側傞丆恎懱偺栻暔偵懳偡傞惗棟揑弴墳忬懺偱偁傞丅
乵夝丂愢乶丂懴惈偼丆僆僺僆僀僪偵尷傜偢挿婜娫栻暔偵敇業偝傟傞偙偲偵傛偭偰惗偠傞惗懱偺惗棟妛揑側揔墳忬懺偱偁傞丅懴惈偑惗偠偰偄傞偐偳偆偐偼丆摨偠岠壥偑摼傜傟傞偙偲偑尒崬傑傟傞偵傕偐偐傢傜偢丆栻暔傪憹検偟偰傕摨偠岠壥偑擣傔傜傟側偔側傞偙偲偱敾抐偡傞丅懴惈宍惉偼栻暔偺栻棟嶌梡偛偲偵堎側傞丅儌儖僸僱偺応崌丆埆怱丒歲揻丆柊婥側偳偵偼懴惈傪宍惉偡傞偑丆曋旈傗弅摰偵偼懴惈傪宍惉偟側偄丅
僆僺僆僀僪偺応崌丆捝傒偺尨場偲側偭偰偄傞庮釃偺憹戝偑側偄偵傕偐偐傢傜偢捔捝岠壥偑庛偔側傞偙偲丆偁傞偄偼丆庮釃偺憹戝偵敽偭偨捝傒偵懳偟偰僆僺僆僀僪傪憹検偟偰傕偦傟偵尒崌偭偨捔捝岠壥偑摼傜傟側偄偙偲偱敾抐偝傟傞丅
亂嶲峫暥專亃
1乯 Savage SR, Joranson DE, Covington EC, et al. Definitions related to the medical use of opioids丗 evolution towards universal agreement. J Pain Symptom Manage 2003丟26丗655-67
2乯 栘郪媊擵丆怷揷払栫丂曇丏梡岅偲夝愢丏擔杮堛巘夛丂娔丏2008 擭斉偑傫娚榓働傾僈僀僪僽僢僋丆搶嫗丆惵奀幮丆2008丆p4
3乯 Portenoy RK. Chronic opioid therapy in nonmalignant pain. J Pain Symptom Manage 1990丟 5丗S46-62
4乯 WHO. Technical Report Series, No.丂915丗2003
5乯 拞崻堯暥丆壀嶈桾巑丆摗尨柇巕丂栿丏ICD-10丗惛恄媦傃峴摦偺忈奞丆搶嫗丆堛妛彂堾丆2003
6乯 崅嫶嶰榊丆戝栰丂桾丆愼栴弐岾丂栿丏DSM-嘩-TR丗惛恄幘姵偺暘椶偲恌抐偺庤堷丆搶嫗丆堛妛彂堾丆2003
❷ 栻棟妛揑婎斦
栻棟妛揑尋媶偼丆墛徢惈醬捝儌僨儖摦暔傗恄宱忈奞惈醬捝儌僨儖摦暔傪偑傫醬捝偺堦晹傪斀塮偟偨儌僨儖偲傒側偟偰峴傢傟偰偄傞丅
1 乯惛恄埶懚
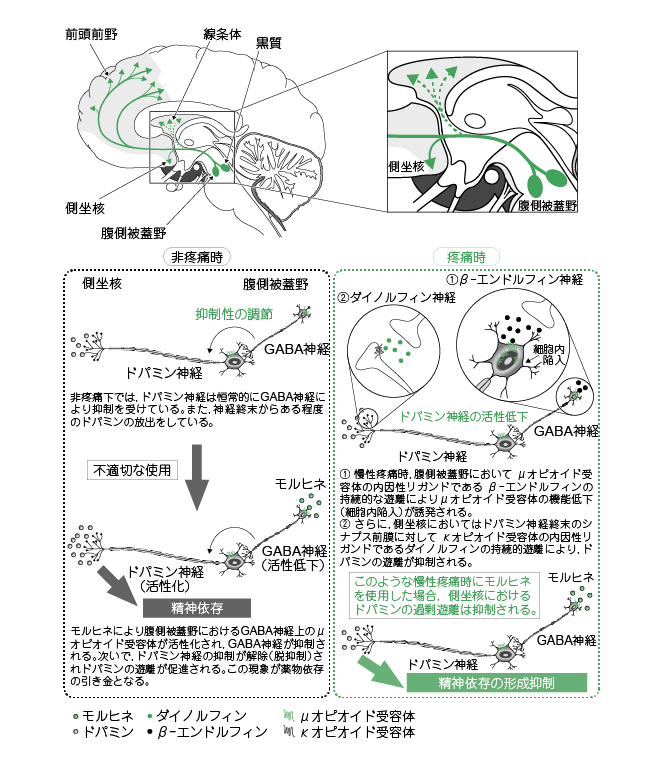
婎慴尋媶偵偍偗傞僆僺僆僀僪偺惛恄埶懚偺昡壙偵偼丆乽忦審偯偗応強歯岲惈帋尡乮conditioned place preference 朄丟CPP 朄乯乿傪梡偄偰丆僆僺僆僀僪偵傛傝桿敪偝傟傞曬廣岠壥傪掕検壔偟偰偄傞丅墛徢惈側傜傃偵恄宱忈奞惈醬捝儌僨儖儅僂僗偵偍偗傞儌儖僸僱桿敪曬廣岠壥傪偙偺CPP 朄偵廬偭偰惛恄埶懚傪昡壙偟偨尋媶偵傛傟偽丆墛徢惈醬捝儌僨儖摦暔偵偍偗傞儌儖僸僱桿敪曬廣岠壥偼傎傏姰慡偵梷惂偝傟丆傑偨丆恄宱忈奞惈醬捝儌僨儖摦暔偵偍偄偰傕儌儖僸僱偺慡恎搳梌偁傞偄偼擼幒撪搳梌偵傛偭偰桿敪偝傟傞曬廣岠壥偼慡偔擣傔傜傟側偄偙偲偑曬崘偝傟偰偄傞丅偝傜偵丆嵟嬤傛傝惛搙偺崅偄惛恄埶懚偺昡壙朄偱偁傞栻暔偺惷柆撪帺屓搳梌朄傪梡偄丆儌儖僸僱丆僼僃儞僞僯儖側偳偺惛恄埶懚偑恄宱忈奞惈醬捝儌僨儖儔僢僩偱梷惂偝傟傞偙偲傕曬崘偝傟偰偄傞丅椪彴宱尡忋丆偑傫醬捝帯椕偵偍偄偰僆僺僆僀僪偺惛恄埶懚偑栤戣偵側傜側偄偙偲偑抦傜傟偰偍傝丆摦暔帋尡偵偍偄偰摨條偺偙偲偑幚徹偝傟丆偝傜偵徻嵶側婡彉傕柧傜偐偵偝傟偰偄傞丅
僆僺僆僀僪偺惛恄埶懚敪尰乮恾3乯偵偼丆拞擼曈墢僪僷儈儞恄宱宯仏偺妶惈壔偑廳梫側栶妱傪壥偨偟偰偄傞丅帠幚丆旕醬捝壓偱偼拞擼曈墢僪僷儈儞恄宱宯偺婲巒妀偱偁傞暊懁旐奧栰偵搳幩偟偰偄傞兞-aminobutyric acid乮GABA乯夘嵼恄宱忋偵懡偔暘晍偟偰偄傞兪僆僺僆僀僪庴梕懱偑儌儖僸僱偵傛傝妶惈壔偝傟丆梷惂惈GABA 夘嵼恄宱偑梷惂偝傟傞丅偦偺寢壥丆扙梷惂婡峔偵傛傝拞擼曈墢僪僷儈儞恄宱宯偼妶惈壔偝傟丆搳幩愭偱偁傞慜擼曈墢晹偺懁嵖妀偵偍偄偰僪僷儈儞梀棧偑懀恑偝傟丆惛恄埶懚偑宍惉偝傟傞丅堦曽丆內僆僺僆僀僪庴梕懱偼庡偵懁嵖妀椞堟偵崅枾搙偵暘晍偟偰偍傝丆妶惈壔偝傟傞偲懁嵖妀偵偍偗傞僪僷儈儞梀棧傪梷惂偡傞偨傔偵寵埆岠壥傪敪尰偡傞丅
仏丗拞擼曈墢僪僷儈儞恄宱宯
恄宱揱払暔幙偲偟偰僪僷儈儞傪棙梡偡傞僪僷儈儞恄宱宯偺堦偮丅擼姴偺暊懁旐奧栰偐傜丆擼偺曈墢宯偵幉嶕廔枛傪搳幩偡傞丅夣偺忣摦傗栻暔埶懚側偳偺恄宱婡峔側偳偵娭梌丅
枬惈墛徢惈醬捝壓偵偍偗傞儌儖僸僱偺惛恄埶懚偺宍惉梷惂偼兪丆兟丆內僆僺僆僀僪庴梕懱偺偦傟偧傟偺漢峈栻偺側偐偱丆內僆僺僆僀僪庴梕懱漢峈栻偺張抲偵傛偭偰偺傒徚幐偡傞偙偲偐傜丆墛徢惈醬捝壓偱偼撪場惈內僆僺僆僀僪恄宱宯偺槾恑偑婲偒偰偄傞偲峫偊傜傟傞丅慜弎偺偲偍傝丆儌儖僸僱偼懁嵖妀椞堟偱偺僪僷儈儞偺挊柧側梀棧傪堷偒婲偙偟偰惛恄埶懚傪桿敪偡傞偑丆枬惈墛徢惈醬捝儌僨儖儔僢僩偺懁嵖妀偵偍偗傞儌儖僸僱桿敪僪僷儈儞梀棧偼丆旕醬捝壓偺儔僢僩偲斾妑偟偰桳堄側梷惂偑擣傔傜傟偨丅偙傟傜偺抦尒偐傜丆枬惈墛徢惈醬捝壓偱偼丆懁嵖妀偵偍偗傞內僆僺僆僀僪恄宱宯偺槾恑偵傛傝丆儌儖僸僱偵傛傞拞擼曈墢僪僷儈儞恄宱宯偺妶惈壔偵傛傞僪僷儈儞梀棧偑梷惂偝傟丆儌儖僸僱偺惛恄埶懚宍惉偑梷惂偝傟傞偲偄偆婡彉偑憐掕偝傟偰偄傞丅
堦曽丆恄宱忈奞惈醬捝儌僨儖偵偍偗傞儌儖僸僱偺惛恄埶懚偺宍惉梷惂偵偼丆內僆僺僆僀僪恄宱宯偑晹暘揑偵偟偐娭傢偭偰偄側偄偙偲偑帵偝傟偰偄傞丅恄宱忈奞惈醬捝偱偼丆暊懁旐奧栰偵搳幩偟偰偄傞兪僆僺僆僀僪庴梕懱偺撪場惈儕僈儞僪仏偱偁傞兝-僄儞僪儖僼傿儞娷桳恄宱偑妶惈壔偝傟丆兝-僄儞僪儖僼傿儞偺梀棧偑帩懕揑偵惗偠傞偨傔丆梷惂惈GABA 夘嵼恄宱忋偵暘晍偟偰偄傞兪僆僺僆僀僪庴梕懱偺扙姶嶌丒婡擻掅壓偑堷偒婲偙偝傟傞偲峫偊傜傟傞丅偙傟傜偺寢壥偐傜丆恄宱忈奞惈醬捝壓偱偼拞擼曈墢僪僷儈儞恄宱宯偑儌儖僸僱側偳偺僆僺僆僀僪偱妶惈壔偝傟偵偔偔側傝丆惛恄埶懚偺宍惉偑梷惂偝傟傞偲憐掕偝傟傞乮恾3乯
仏丗撪場惈儕僈儞僪
庴梕懱傗峺慺偵寢崌偟丆惗暔妶惈傪堷偒婲偙偡暔幙乮儕僈儞僪乯偺偆偪丆摿偵懱撪偱嶻惗偝傟偨暔幙傪巜偡丅
恾3丂枬惈醬捝壓偵偍偗傞僆僺僆僀僪偺惛恄埶懚晄宍惉婡峔
2 乯恎懱埶懚
墛徢惈醬捝儌僨儖摦暔偱儌儖僸僱偺恎懱埶懚傪専摙偟偨尋媶偱偼丆墛徢惈醬捝壓偵偍偗傞儌儖僸僱偺棧扙徢岓偑旕醬捝壓偲斾妑偟偰丆桳堄偵梷惂偝傟偰偄傞丅偝傜偵丆墛徢惈醬捝壓偱傕媫寖側媥栻偱偼庛偄棧扙徢岓偑擣傔傜傟傞偑丆儌儖僸僱偺搳梌検傪慟尭偟偨応崌丆旕醬捝壓偱偼庛偄棧扙徢岓傪帵偡傕偺偺丆墛徢惈醬捝壓偱偼慡偔棧扙徢岓傪帵偝側偄偙偲偑柧傜偐偵偝傟偰偄傞丅
偙偺傛偆側墛徢惈醬捝壓偱偺恎懱埶懚宍惉梷惂婡峔偵娭偡傞専摙偑峴傢傟丆內僆僺僆僀僪庴梕懱偺撪場惈儕僈儞僪偱偁傞僟僀僲儖僼傿儞偼儌儖僸僱埶懚摦暔偵偍偗傞棧扙徢岓偺敪尰傪梷惂偡傞偙偲丆偝傜偵丆內僆僺僆僀僪庴梕懱漢峈栻偑儌儖僸僱偺恎懱埶懚宍惉傪憹嫮偡傞偙偲偑曬崘偝傟偰偄傞丅偟偨偑偭偰丆枬惈墛徢惈醬捝壓偵偍偗傞儌儖僸僱偺恎懱埶懚偺宍惉梷惂偵偼撪場惈內僆僺僆僀僪恄宱宯偺妶惈壔偑娭梌偟偰偄傞偲峫偊傜傟傞丅
3 乯懴惈乮捔捝懴惈乯
惓忢摦暔偵懳偡傞僆僺僆僀僪偺枬惈搳梌偵傛傝捔捝懴惈偑宍惉偝傟傞偙偲偼偁傑傝偵傕桳柤側尰徾偱偁傞丅堦曽丆墛徢惈醬捝傗恄宱忈奞惈醬捝儅僂僗傪梡偄偨専摙偱偼丆僆僺僆僀僪偺捔捝岠壥偼斀暅搳梌偱傕惓忢摦暔偵斾傋偰斾妑揑堐帩偝傟偰偍傝丆捔捝懴惈偼庛偄偲峫偊傜傟傞丅奺僆僺僆僀僪偵傛傞捔捝懴惈偺宍惉掱搙偵偼偁傞掱搙偺嵎偑偁傞傕偺偺丆僆僺僆僀僪偺夁検搳梌偱偼柧妋側捔捝懴惈傪宍惉偡傞偙偲偐傜揔愗側捔捝梡検傪慖戰偡傞偙偲偑廳梫偱偁傞丅
乮楅栘丂曌乯
亂嶲峫暥專亃
1乯 Suzuki T, Kishimoto Y, Misawa M, et al. Role of the kappa-opioid system in the attenuation of the morphine-induced place preference under chronic pain. Life Sci 1999丟64丗1-7
2乯 Narita M, Kishimoto Y, Ise Y, et al. Direct evidence for the involvement of the mesolimbic kappa-opioid system in the morphine-induced rewarding effect under an inflammatory pain- like state. Neuropsychopharmacology 2005丟30丗111-8
3乯 Petraschka M, Li S, Gilbert TL, et al. The absence of endogenous beta-endorphin selectively blocks phosphorylation and desensitization of mu opioid receptors following partial sciatic nerve ligation. Neuroscience 2007丟146丗1795-807
4乯 Martin TJ, Kim SA, Buechler NL, et al. Opioid self-administration in the nerve-injured rat丗 relevance of antiallodynic effects to drug consumption and effects of intrathecal analgesics. Anesthesiology 2007丟106丗312-22
❹ 椪丂彴
惛恄埶懚丆恎懱埶懚丆懴惈偵娭偡傞椪彴揑偵廳梫側揰偼埲壓偺偙偲偱偁傞丅
1 乯惛恄埶懚
偑傫姵幰偺捝傒偵懳偟偰僆僺僆僀僪傪挿婜娫巊梡偟偰傕惛恄埶懚偼傑傟偱偁傞丅偟偐偟丆暔幙埶懚偺婛墲偑偁傞姵幰偺応崌丆旕偑傫醬捝偵懳偡傞巊梡偺応崌傪娷傔丆惛恄埶懚傪媈偆峴摦偑傒傜傟偨応崌偵偼丆惛恄堛妛揑側昡壙傪娷傔偰丆捝傒偵懳偡傞僆僺僆僀僪搳梌偺懨摉惈傪嵞専摙偡傞丅惛恄埶懚偵娭偡傞愱栧揑抦幆傪桳偟偰偄傞惛恄壢堛側偳偺愱栧壠偵憡択偡傞偙偲偑朷傑偟偄丅
2 乯恎懱埶懚
恎懱埶懚偼偑傫醬捝偑懚嵼偟丆僆僺僆僀僪偑宲懕搳梌偝傟傞尷傝偼栤戣偵側傜側偄丅椪彴忋栤戣偲側傞偺偼丆宱岥愛庢偑偱偒側偔側傝宱岥搳梌偟偰偄偨僆僺僆僀僪偑撪暈偱偒側偔側傞側偳媫偵拞抐偟偨応崌丆岆偭偰搳梌検傪嬌抂偵尭検偟偨応崌丆僆僺僆僀僪僗僀僢僠儞僌偵敽偄戝検偺僆僺僆僀僪傪堦搙偵懠偺僆僺僆僀僪偵曄峏偟偨応崌偵丆棧扙徢岓孮傪惗偠摼傞丅椺偊偽丆宱岥儌儖僸僱彊曻惈惢嵻偐傜僼僃儞僞僯儖揬晅嵻傊僗僀僢僠儞僌偟偨応崌丆堦帪揑側壓棢徢忬傪掓偡傞偙偲偑偁傞偑丆偙傟偼儌儖僸僱恎懱埶懚偵敽偭偨棧扙徢岓孮偺堦偮偲峫偊傜傟傞丅僆僺僆僀僪偺棧扙徢岓孮偼丆搳梌偝傟偰偄偨僆僺僆僀僪傪彮検搳梌偡傞偙偲偱徢忬偼徚幐偡傞丅棧扙徢岓孮偺敪尰梊杊偲偟偰丆媫偵僆僺僆僀僪傪拞抐偣偢丆尭検偑昁梫側応崌偵偼彊乆偵尭検偡傞偙偲偑昁梫偱偁傞丅
3 乯懴丂惈
懴惈偼丆捝傒偺昡壙傪廫暘偵峴偄丆揔愗側検偺僆僺僆僀僪傪搳梌偟偰偄傟偽栤戣偵側傞偙偲偼彮側偄丅梊杊偲偟偰偼丆僆僺僆僀僪偺巊梡検傪偄偨偢傜偵憹検偟側偄傛偆偵偟丆捝傒偵墳偠偨帯椕傪暪梡偡傞乮NSAIDs丆曻幩慄帯椕丆恄宱僽儘僢僋丆捔捝曗彆栻丆旕栻暔揑庤抜側偳乯偙偲偑廳梫偱偁傞丅憹検偵尒崌偆捔捝岠壥偑擣傔傜傟側偄応崌偵偼丆僆僺僆僀僪僗僀僢僠儞僌丆僆僺僆僀僪埲奜偺捔捝庤抜側偳傪専摙偡傞丅
乮晊埨巙榊丆楅栘丂曌乯
俀
旕僆僺僆僀僪捔捝栻
侾丏旕僗僥儘僀僪惈徚墛捔捝栻乮NSAIDs乯
❶ 栻棟妛揑摿挜
NSAIDs 偼僗僥儘僀僪峔憿埲奜偺峈墛徢嶌梡丆夝擬嶌梡丆捔捝嶌梡傪桳偡傞栻暔偺憤徧偱偁傞丅
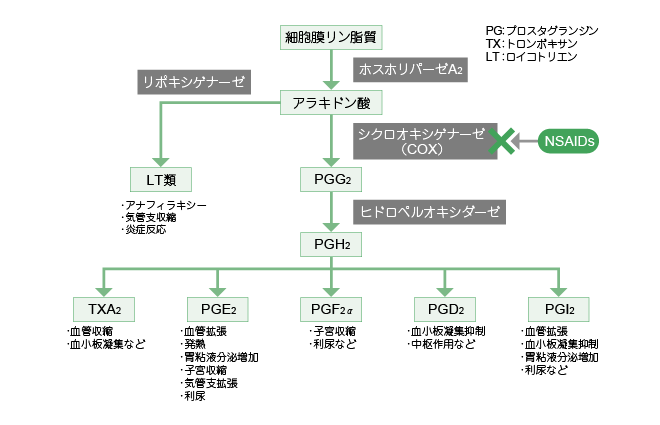
乵嶌梡婡彉乶丂NSAIDs 偺庡側岠壥偼丆墛徢偑偁傞嬊強偵偍偗傞僾儘僗僞僌儔儞僕儞乮prostaglandin丟PG乯偺嶻惗慾奞偱偁傞丅慻怐偑懝彎偝傟傞偲丆儂僗儂儕僷乕僛A2 偵傛傝丆嵶朎枌偺儕儞帀幙偐傜傾儔僉僪儞巁偑梀棧偝傟傞丅梀棧偝傟偨傾儔僉僪儞巁偼僔僋儘僆僉僔僎僫乕僛乮cyclooxygenase丟COX乯傗儁儖僆僉僔僟乕僛傪娷傓PGH乮prostaglandin H乯崌惉峺慺暋崌懱偺婎幙偲側傝丆PGG2丆PGH2傊偲曄姺偝傟傞丅偝傜偵奺慻怐偵摿堎揑側PG 崌惉峺慺偵傛傝PGE2乮prostaglandin E2乯側偳庬乆偺壔妛揱払暔幙偑崌惉偝傟丆懝彎慻怐傊曻弌偝傟傞丅PG 帺懱偵敪捝嶌梡偼側偄偑丆僽儔僕僉僯儞側偳偺敪捝暔幙偺醬捝鑷抣傪掅壓偝偣傞丅傑偨丆嬊強偱偺寣棳憹壛嶌梡傗寣娗摟夁惈偺槾恑丆敀寣媴偺怹弫憹壛側偳丆墛徢傪憹嫮偝偣傞嶌梡傪桳偡傞丅偟偨偑偭偰丆NSAIDs 偼梀棧偝傟偨傾儔僉僪儞巁偐傜PG 傪崌惉偡傞宱楬偺棩懍峺慺偱偁傞僔僋儘僆僉僔僎僫乕僛偺摥偒傪慾奞偡傞偙偲偵傛傝峈墛徢丒捔捝嶌梡傪敪婗偡傞乮恾4乯丅
傑偨丆敪擬帪偵偼庬乆偺僒僀僩僇僀儞偺嶻惗偑懀恑偝傟丆帇彴壓晹偵偁傞懱壏挷愡拞悤偵偍偗傞PGE2偺崌惉傪憹壛偝偣丆懱壏傪忋徃偝偣傞傛偆偵帇彴壓晹偵摥偒偐偗傞丅NSAIDs 偼敪擬帪偵嶻惗偝傟傞PGE2偺崌惉傪慾奞偡傞偙偲偱丆夝擬嶌梡傪傕偨傜偡丅
乵嶌梡帪娫乶丂傾僗僺儕儞偼僔僋儘僆僉僔僎僫乕僛偺妶惈晹埵傪傾僙僠儖壔偟偰晄壜媡揑偵慾奞偡傞丅偙偺偨傔傾僗僺儕儞偺嶌梡帪娫偼庬乆偺昗揑慻怐偱僔僋儘僆僉僔僎僫乕僛偑敪尰偟丆抲偒姺傢傞懍搙偲娭學偡傞丅懠偺NSAIDs 偼僔僋儘僆僉僔僎僫乕僛偺妶惈拞怱偵偍偄偰傾儔僉僪儞巁偲壜媡揑偵漢峈偟偰丆偦偺摥偒傪慾奞偡傞偨傔偵丆嶌梡帪娫偼栻暔偺寣拞擹搙敿尭婜偵埶懚偡傞丅
乵僔僋儘僆僉僔僎僫乕僛傾僀僜僓僀儉慖戰惈乶丂僔僋儘僆僉僔僎僫乕僛偵偼丆COX-1 偲COX-2 偺2 偮偺傾僀僜僓僀儉偑懚嵼偡傞丅COX-1 偼戝晹暘偺惓忢嵶朎傗慻怐偵掕忢揑偵敪尰偟丆恎懱婡擻偺堐帩偵娭梌偟偰偄傞丅堦曽丆COX-2 偼墛徢偵敽偄僒僀僩僇僀儞傗墛徢儊僨傿僄乕僞乕偵傛偭偰桿摫偝傟傞偑丆恡憻傗擼偺摿掕偺椞堟偱偼掕忢揑偵敪尰偟偰偄傞丅堓擲枌偺忋旂嵶朎偱偼COX-1 偑掕忢揑偵敪尰偟偰偍傝丆嵶朎曐岇岠壥傪傕偮PG 偺嶻惗偵娭傢偭偰偄傞丅崙撪偱棙梡壜擻側NSAIDs 偼偄偢傟傕掱搙偺嵎偼偁傞傕偺偺丆COX-1 偍傛傃COX-2 偺偳偪傜偺妶惈傕梷惂偡傞丅慖戰揑COX-2 慾奞栻偲偟偰僙儗僐僉僔僽偑偁傝丆斾妑揑COX-2 慾奞偺慖戰惈偑崅偄傕偺偵僄僩僪儔僋丆儊儘僉僔僇儉偑偁傞丅
恾4丂傾儔僉僪儞巁偺戙幱宱楬
❷ 暃嶌梡
NSAIDs 偺暃嶌梡偼嫟捠偟偰傒傜傟傞傕偺偲丆屄乆偺NSAIDs 偵摿堎揑偵傒傜傟傞傕偺偑偁傞丅嫟捠偡傞庡側暃嶌梡傪昞10 偵帵偡丅
| 晹埵摍 | 徢丂忬 | 峫偊傜傟傞婡彉偺堦晹 旛峫 |
|---|---|---|
堓丂挵 |
暊捝丆埆怱丆怘梸晄怳丆堓傃傜傫丒 捵釃丆堓挵娗弌寣丆慂岴丆壓棢 |
堓擲枌忋旂嵶朎偱偺COX-1 偺慾奞偵傛傞PGI2丆PGE2側偳偺尭彮 |
恡丂憻 |
悈丒揹夝幙挋棷丆崅K 寣徢丆晜庮丆 娫幙惈恡墛丆僱僼儘乕僛徢岓孮 |
恡偵偍偗傞COX 偺慾奞偵傛傞PG 尭彮偵敽偆恡寣棳検偲巺媴懱鄅夁懍搙偺尭彮 |
娞丂憻 |
娞婡擻専嵏抣堎忢丆娞晄慡 |
僕僋儘僼僃僫僋丆僗儕儞僟僋側偳摿偵拲堄 |
寣彫斅 |
寣彫斅妶惈壔慾奞丆弌寣偺婋尟憹壛 |
寣彫斅偱偺COX-1 偺慾奞偵傛傞TXA2偺尭彮偵敽偆寣彫斅嬅廤擻偺掅壓 |
晄懴乮夁晀乯徢 |
寣娗乮塣摦乯恄宱惈旲墛丆寣娗晜庮丆歜懅丆 偠傫傑怾丆婥娗巟歜懅丆挭峠丆掅寣埑丆僔儑僢僋 |
COX 偺慾奞偵敽偆LT 椶偺崌惉憹壛摍 |
拞悤恄宱宯 |
摢捝丆傔傑偄丆嶖棎丆梷偆偮丆偗偄傟傫偺鑷抣掅壓 |
偗偄傟傫偺鑷抣掅壓丗擼撪偱偺GABA 偺庴梕懱寢崌慾奞 |
旂晢丒擲枌 |
旂怾丆岝夁晀徢乮摿偵僼僃僯儖僾儘僺僆儞巁宯乯丆旂晢擲枌娽徢岓孮丆拞撆惈昞旂夡巰徢 | 岝撆惈 柶塽丒傾儗儖僊乕揑斀墳側偳 |
擠怭帪 |
擠怭婜娫偺墑挿丆暘曍慾奞 戀帣偺摦柆娗暵嵔 |
COX 偺慾奞偵敽偆PGE2丆PGF2α偺尭彮 |
1 乯堓挵忈奞
徚壔惈捵釃宍惉偼丆僿儕僐僶僋僞乕丒僺儘儕姶愼傗傾儖僐乕儖偺夁忚愛庢丆僐儖僠僐僗僥儘僀僪傗峈嬅屌栚揑偺掅梡検傾僗僺儕儞暪梡側偳偺擲枌懝彎場巕偵傛傝婋尟搙偑崅傑傞丅NSAIDs 偵傛傞堓挵忈奞偵偼丆堓擲枌忋旂嵶朎偵偍偗傞COX-1 慾奞偵傛偭偰堷偒婲偙偝傟傞擲枌嵶朎曐岇岠壥傪傕偮PGI2丆PGE2側偳偺尭彮偑怺偔娭傢偭偰偄傞丅慖戰揑COX-2 慾奞栻偼廬棃偺NSAIDs 傛傝堓捵釃敪徢偺昿搙偑掅偄偲偝傟偰偄傞丅傑偨宱岥搳梌帪偵偼丆NSAIDs 偑堓擲枌偵捈愙愙怗偡傞偙偲偱偺嬊強巋寖傕娭梌偟偰偄傞丅
堓捵釃偺梊杊栻偲偟偰丆僾儘僗僞僌儔儞僕儞惢嵻乮儈僜僾儘僗僩乕儖乯丆僾儘僩儞億儞僾慾奞栻丆崅梡検偺H2庴梕懱漢峈栻側偳偑巊梡偝傟偰偄傞丅
2 乯恡婡擻忈奞
偆偭寣惈怱晄慡丆暊悈傪敽偆娞峝曄丆枬惈恡幘姵丆傑偨偼弞娐寣棳検偑尭彮偟偰偄傞姵幰偱偼恡寣棳検偲巺媴懱鄅夁懍搙偑尭彮偟媫惈恡晄慡傪婲偙偡偙偲偑偁傞丅恡婡擻忈奞偑偁傞姵幰傗崅楊幰偵搳梌偡傞嵺偼丆廫暘側拲堄傪偡傞昁梫偑偁傞丅
3 乯寣彫斅丆怱寣娗宯忈奞
妶惈壔偟偨寣彫斅偱偼COX-1 偺攠夘偵傛傝僩儘儞儃僉僒儞A2乮TXA2乯偑惗惉偝傟寣愷宍惉傪懀恑偡傞丅懳徠揑偵撪旂嵶朎偱偼COX-2 偺攠夘偵傛傝PGI2偑惗惉偝傟峈寣愷嶌梡傪帵偡丅
NSAIDs 偼僔僋儘僆僉僔僎僫乕僛傪慾奞偟丆TXA2偺寣彫斅宍惉傪梷惂偡傞偨傔寣彫斅婡擻偑忈奞偝傟丆弌寣孹岦偑尰傟傞偙偲偑偁傞丅寣彫斅偱偼庡偵COX-1 偑敪尰偟偰偄傞偨傔丆慖戰揑COX-2 慾奞栻偱偼寣彫斅婡擻忈奞偑寉尭偝傟傞丅
怱寣娗忈奞偺敪徢憹壛偺儕僗僋偼丆慖戰揑COX-2 慾奞栻偱偁傞僐僉僔僽宯栻嵻偺戝婯柾椪彴帋尡偱柧傜偐偲側偭偨丅COX-2 傪慖戰揑偵慾奞偡傞偨傔丆PGI2惗惉傪慾奞偡傞偑丆TXA2惗惉偵偼塭嬁傪梌偊偢丆峈寣愷偲寣愷宍惉懀恑偺娫偱晄嬒摍傪惗偠寣愷宍惉偵孹偔偲峫偊傜傟偰偄傞丅偟偐偟丆旕慖戰揑側COX 慾奞栻偱偁傞廬棃偺NSAIDs乮傾僗僺儕儞傪彍偔乯偵偍偄偰傕怱寣娗忈奞偺敪徢偑曬崘偝傟偰偍傝丆徻嵶側敪惗婡彉偼晄柧偱偁傞丅
4 乯傾僗僺儕儞晄懴乮夁晀乯徢
傾僗僺儕儞傗偦偺懠偺NSAIDs 偵夁晀偱丆寣娗晜庮丆慡恎惈偠傫傑怾丆婥娗巟歜懅丆岮摢晜庮丆僔儑僢僋側偳偺偝傑偞傑側徢忬傪帵偡応崌偑偁傞丅傾僗僺儕儞晄懴乮夁晀乯徢偺徢忬偼傾僫僼傿儔僉僔乕偲傕椶帡偟偰偄傞偑丆柶塽斀墳偱偼側偔僔僋儘僆僉僔僎僫乕僛偺慾奞偑娭傢偭偰偄傞偲峫偊傜傟偰偄傞丅
俀丏傾僙僩傾儈僲僼僃儞
❶ 栻棟妛揑摿挜
傾僙僩傾儈僲僼僃儞乲暿柤乮崙嵺堦斒柤乯丗僷儔僙僞儌乕儖乴偼傾僗僺儕儞偲摨摍偺捔捝丆夝擬嶌梡傪傕偮桳梡側栻暔偱偁傞偑丆峈墛徢嶌梡偼旕忢偵庛偄偲峫偊傜傟偰偄傞丅庡偵拞悤偵嶌梡偟偰捔捝嶌梡傪敪尰偡傞丅徚壔娗丆恡婡擻丆寣彫斅婡擻偵懳偡傞塭嬁偼彮側偄偲峫偊傜傟丆偙傟傜偺忈奞偱NSAIDs 偑巊梡偟偵偔偄応崌偵傕梡偄傞偙偲偑偱偒傞丅
❷ 梡朄丒梡検
廬棃丆傾僙僩傾儈僲僼僃儞偺杮朚偺揧晅暥彂偱偼丆偑傫醬捝傊偺揔墳偼偁傞傕偺偺丆惉恖偱偼傾僙僩傾儈僲僼僃儞1 夞300乣500 mg丆1 擔900乣1,500 mg 傪揔媂憹尭偟偰搳梌偡傞偲側偭偰偄偨丅偟偐偟丆墷暷偍傛傃傾僕傾偺堦晹偱偑傫醬捝偵懳偟偰巊梡偝傟傞傾僙僩傾儈僲僼僃儞偺宱岥搳梌検偼丆1 夞650 mg 傪4 帪娫枅丆傑偨偼1,000 mg 傪6 帪娫枅丆1 擔嵟戝検偼4,000 mg/擔偱偁傝丆杮朚偱傕偑傫醬捝偱偼2,400乣4,000 mg/擔掱搙偑懨摉側捔捝検偲偟偰巊梡偝傟偰偒偨丅
2011 擭1 寧偵杮朚偺傾僙僩傾儈僲僼僃儞偺梡検丒梡朄偑夵掶偝傟丆傾僙僩傾儈僲僼僃儞偲偟偰1 夞300乣1,000 mg 傪宱岥搳梌丆搳梌娫妘偼4乣6 帪娫埲忋丆1 擔嵟戝検偼4,000 mg/擔偲側偭偨丅傾僙僩傾儈僲僼僃儞傪娷傓攝崌嵻偲偺暪梡偵傕拲堄偡傞昁梫偑偁傞丅奀奜偱偼丆2002 擭偐傜傾僙僩傾儈僲僼僃儞偺惷拲塼偑巊梡偝傟偰偒偨偑丆2013 擭傛傝杮朚偱傕巊梡偑壜擻偲側偭偨丅
❸ 暃嶌梡
堦斒揑側搳梌検偱偼暃嶌梡偼婲偙傝偵偔偄偑丆傑傟偵旂晢擲枌娽徢岓孮丆旂怾丆偦偺懠偺傾儗儖僊乕徢忬丆夁晀徢忬丆娞婡擻忈奞丆墿醫側偳偑婲偙傞丅傑偨丆梓棻媴尭彮徢丆娫幙惈攛墛丆娫幙惈恡墛偺曬崘椺偑偁傞丅嵟傕廳撃側媫惈偺暃嶌梡偼丆夁忚搳梌偵傛傞娞嵶朎夡巰偱偁傞丅惉恖偱偼丆1 夞偵150乣250 mg/kg 埲忋偺傾僙僩傾儈僲僼僃儞傪宱岥搳梌偡傞偲娞嵶朎夡巰偑婲偙傞壜擻惈偑偁傝丆500 mg/kg 偱偼崅妋棪偱敪惗偡傞偲曬崘偝傟偰偄傞丅傾儖僐乕儖忢梡幰丆塰梴忬懺偺埆壔丆栻暔戙幱峺慺乮CYP2E1乯傪桿摫偡傞栻暔乮僀僜僯傾僕僪摍乯偲偺暪梡偱偼偦偺儕僗僋偑崅傑傞丅傾僙僩傾儈僲僼僃儞夁忚愛庢帪偺夝撆偵偼傾僙僠儖僔僗僥僀儞偑巊梡偝傟傞丅
乮棿丂宐旤乯
俁
捔捝曗彆栻
侾丏捔捝曗彆栻偺掕媊
乵掕丂媊乶丂庡偨傞栻棟嶌梡偵偼捔捝嶌梡傪桳偟側偄偑丆捔捝栻偲暪梡偡傞偙偲偵傛傝捔捝岠壥傪崅傔丆摿掕偺忬嫷壓偱捔捝岠壥傪帵偡栻暔偱偁傞丅
乵夝丂愢乶丂乽捔捝曗彆栻乿偺掕媊偵偼峀媊偺傕偺偲嫹媊偺傕偺偲偑偁傞丅
峀媊偺傕偺偼丆WHO 曽幃偑傫醬捝帯椕朄傪偼偠傔偲偟偰嵦梡偝傟偰偄傞傕偺偱丆惂揻栻側偳偺暃嶌梡懳張栻傪娷傓丅2000 擭偵岞昞偟偨擔杮娚榓堛椕妛夛偺乽Evidenced-Based Medicine 偵懃偭偨偑傫醬捝帯椕僈僀僪儔僀儞乿偱偼丆捔捝傪栚揑偲偟偰巊梡偡傞傕偺傪戞1 庬捔捝曗彆栻丆偦傟埲奜傪戞2 庬捔捝曗彆栻偲掕媊偟偨丅
杮僈僀僪儔僀儞偱偼丆Lussier 傜偺Oxford Textbook of Palliative Medicine 偺婰嵹傪嶲峫偵丆捔捝曗彆栻偲偟偰忋婰偺傛偆偵嫹媊偺掕媊傪梡偄偨丅
俀丏捔捝曗彆栻偺奣梫
恄宱忈奞惈醬捝傪偼偠傔偲偡傞僆僺僆僀僪掞峈惈偺捝傒偵懳偟偰丆尰嵼丆懡偔偺栻嵻偑捔捝曗彆栻偲偟偰巊梡偝傟偰偄傞偑丆幙偺崅偄椪彴帋尡偼彮側偔丆揔惓側巊梡曽朄偵偮偄偰偼偄傑偩偵妋棫偝傟偰偄側偄丅懷忬醰怾屻恄宱捝丆摐擜昦惈枛徑恄宱忈奞偼丆懳徾偺捝傒偺惈幙偑斾妑揑嬒堦偲峫偊傜傟丆偙傟傜偺旕偑傫惈恄宱忈奞惈醬捝偺帋尡惉愌傪傕偲偵丆偑傫偵傛傞恄宱忈奞惈醬捝偵巊梡偝傟傞偙偲偑懡偄丅捝傒偺婡彉偵婎偯偒帯椕朄傪慖戰偟丆NNT仏1乮number needed to treat乯偑彫偝偔丆NNH仏2乮number needed to harm乯偑戝偒側栻暔傪慖戰偡傞偙偲偑丆恄宱忈奞惈醬捝偵懳偡傞岠壥揑偐偮埨慡側帯椕愴棯偲側傞偑丆慜弎偺偲偍傝丆廫暘側椪彴帋尡偵婎偯偔僨乕僞偑彮側偄偆偊偵丆杮朚偱巊梡偱偒傞栻嵻偼尷傜傟傞丅傑偨尰忬偵偍偄偰偼丆恄宱忈奞惈醬捝偵懳偡傞僾儗僈僶儕儞埲奜丆偦偺傎偲傫偳偑曐尟揔梡奜偺巊梡偲側傞丅
偙傟傜傪傆傑偊偨偆偊偱丆嶲峫偲偟偰昞11 偵捔捝曗彆栻偺搳梌曽朄偺栚埨傪婰嵹偟偨丅
仏1丗NNT乮number needed to treat乯
1 椺偺岠壥傪摼傞偨傔偵偦偺帯椕傪壗恖偺姵幰偵梡偄側偗傟偽側傜側偄偐傪帵偡巜昗丅
仏2丗NNH乮number needed to harm乯
壗恖偺姵幰傪帯椕偡傞偲1 椺偺桳奞徢椺偑弌尰偡傞偐傪帵偡巜昗丅
| 栻嵻暘椶 | 惉暘柤 | 梡朄丒梡検 | 旛峫乮庡側暃嶌梡乯 | ||
|---|---|---|---|---|---|
峈偆偮栻 |
TCA | 傾儈僩儕僾僠儕儞 傾儌僉僒僺儞 僲儖僩儕僾僠儕儞 |
奐巒検丗 10 mg/擔丂PO 乮廇怮慜乯 |
堐帩検丗 10乣75 mg/擔丂PO |
柊婥丆岥妷丆曋旈丆攔擜忈奞丆柖帇側偳 |
| SNRI | 僨儏儘僉僙僠儞 | 奐巒検丗 20 mg/擔丂PO 乮挬怘屻乯 |
堐帩検丗 40乣60 mg/擔丂PO |
埆怱乮奐巒弶婜偵懡偄乯丆怘梸晄怳丆摢捝丆晄柊丆晄埨丆嫽暠側偳 | |
| SSRI | 僷儘僉僙僠儞 | 奐巒検丗 20 mg乮崅楊幰偼10 mg乯/擔丂PO |
|||
| 僼儖儃僉僒儈儞 | 奐巒検丗25 mg/擔丂PO | ||||
峈偗偄傟傫栻 |
僾儗僈僶儕儞 | 奐巒検丗 50乣150 mg/擔丂PO 乮廇怮慜傑偨偼暘2乯 |
堐帩検丗 300乣600 mg/擔丂PO |
柊婥丆傆傜偮偒丆傔傑偄丆 枛徑惈晜庮側偳 |
|
| 僈僶儁儞僠儞 | 奐巒検丗 200 mg/擔丂PO 乮廇怮慜乯 |
堐帩検丗 2,400 mg/擔丂PO |
柊婥丆傆傜偮偒丆傔傑偄丆 枛徑惈晜庮側偳 |
||
| 僶儖僾儘巁 | 奐巒検丗 200 mg/擔丂PO 乮廇怮慜乯 |
堐帩検丗 400乣1,200 mg/擔丂PO |
柊婥丆埆怱丆娞婡擻忈奞丆 崅傾儞儌僯傾寣徢側偳 |
||
| 僼僃僯僩僀儞 | 堐帩検丗150乣300 mg/擔丂PO乮暘3乯 | 柊婥丆塣摦幐挷丆埆怱丆娞忈奞丆旂晢徢忬側偳 | |||
| 僋儘僫僛僷儉 | 奐巒検丗 0.5 mg/擔丂PO 乮廇怮慜乯 |
堐帩検丗 1乣2 mg/擔丂PO |
傆傜偮偒丆柊婥丆傔傑偄丆 塣摦幐挷側偳 |
||
峈晄惍柆栻 |
儊僉僔儗僠儞 | 奐巒検丗 150 mg/擔丂PO乮暘3乯 |
堐帩検丗 300 mg/擔丂PO乮暘3乯 |
埆怱丆怘梸晄怳丆暊捝丆 堓挵忈奞側偳 |
|
| 儕僪僇僀儞 | 奐巒検丗 5 mg/kg/擔 CIV丆CSC |
堐帩検丗 5乣20 mg/kg/擔丂CIV丆CSC |
晄惍柆丆帹柭丆嫽暠丆 偗偄傟傫丆柍姶妎側偳 |
||
NMDA 庴梕懱 |
働僞儈儞 | 奐巒検丗 0.5乣1 mg/kg/擔 CIV丆CSC |
堐帩検丗 100乣300 mg/擔丂CIV丆CSC |
柊婥丆傆傜偮偒丆傔傑偄丆埆柌丆埆怱丆偣傫栂丆偗偄傟傫乮擼埑槾恑乯側偳 | |
拞悤惈嬝抩娚栻 |
僶僋儘僼僃儞 | 奐巒検丗 10乣15 mg/擔丂PO 乮暘2乣3乯 |
堐帩検丗 15乣30 mg/擔丂PO |
柊婥丆摢捝丆寫懹姶丆 堄幆忈奞側偳 |
|
僐儖僠僐 |
儀僞儊僞僝儞 僨僉僒儊僞僝儞 |
①慟尭朄 丂奐巒検丗4乣8 mg/擔乮暘1乣2丗梉曽埲崀偺搳梌傪旔偗傞乯 丂堐帩検丗0.5乣4 mg/擔 ②慟憹朄 丂奐巒検丗0.5 mg/擔 丂堐帩検丗4 mg/擔 |
崅寣摐丆崪慹偟傚偆徢丆 徚壔惈捵釃丆堈姶愼徢側偳 |
||
儀儞僝僕傾僛僺 |
僕傾僛僷儉 | 2乣10 mg/夞丆1 擔3乣4 夞 | 傆傜偮偒丆柊婥丆塣摦幐挷 側偳 |
||
Bone-modifying |
僝儗僪儘儞巁 | 4 mg 傪15 暘埲忋偐偗偰DIV丆3乣4 廡枅 | 妠崪夡巰丆媫惈恡晄慡丆 偆偭寣惈怱晄慡丆敪擬丆娭愡捝側偳 |
||
| 僨僲僗儅僽 | 120 mg 傪SC丆4 廡偵1 夞 | 掅僇儖僔僂儉寣徢丆妠崪夡巰丒妠崪崪悜墛側偳 | |||
偦偺懠 |
僆僋僩儗僆僠僪 | 0.2乣0.3 mg/擔丂CSC 傑偨偼SC乮0.1 mg×3 夞乯 | 拲幩晹埵偺峝寢丒敪愒丒巋寖姶側偳 | ||
| 僽僠儖僗僐億儔儈儞 | 奐巒検丗10乣20 mg/擔丂CSC丆CIV | 怱湩槾恑丆岥妷丆娽偺挷愡忈奞側偳 | |||
PO丗宱岥丆CIV丗帩懕惷拲丆SC丗旂壓拲丆CSC丗帩懕旂壓拲丆DIV丗揰揌惷拲
TCA丗嶰娐宯峈偆偮栻丆SNRI丗僙儘僩僯儞丒僲儖傾僪儗僫儕儞嵞庢傝崬傒慾奞栻丆SSRI丗慖戰揑僙儘僩僯儞嵞庢傝崬傒慾奞栻
俁丏奺捔捝曗彆栻偺摿挜
❶ 栻峈偆偮栻
乵嶌梡婡彉丒摿挜乶丂拞悤恄宱宯偺僙儘僩僯儞丆僲儖傾僪儗僫儕儞嵞庢傝崬傒傪慾奞偟丆壓峴惈梷惂宯傪晩妶偡傞偙偲偵傛偭偰捔捝岠壥傪敪婗偡傞丅捔捝岠壥偺敪尰偼丆捠忢偺峈偆偮嶌梡偑敪尰偡傞偲偝傟偰偄傞廡扨埵傛傝傕憗偔丆搳梌奐巒1 廡娫埲撪偵岠壥敪尰偟丆偐偮丆偆偮昦偺帯椕検傛傝傕掅梡検偱峈偆偮嶌梡傪帵偝偢偵捔捝岠壥偑擣傔傜傟傞丅
慖戰揑僙儘僩僯儞嵞庢傝崬傒慾奞嵻乮selective serotonin reuptake inhibitor丟 SSRI乯傛傝丆僙儘僩僯儞偲僲儖傾僪儗僫儕儞偺椉曽偺嶌梡傪偁傢偣傕偮SNRI乮serotonin noradrenaline reuptake inhibitor丟SNRI乯偺傎偆偑捔捝曗彆栻偲偟偰桳梡側壜擻惈偑帵嵈偡傞抦尒偑偁傞偑丆尰嵼偺偲偙傠堦抳偟偨尒夝偼摼傜傟偰偄側偄丅
乵暃嶌梡乶丂傾儈僩儕僾僠儕儞側偳偺嶰娐宯峈偆偮栻偱偼丆柊婥丆峈僐儕儞嶌梡乮岥撪姡憞丆曋旈丆攔擜忈奞丆柖帇側偳乯丆婲棫惈掅寣埑丆偣傫栂偑傒傜傟傞丅廳撃側暃嶌梡偲偟偰偼怱撆惈偑偁傝丆捔捝岠壥傪帵偡搳梌検偱偼傑傟偱偁傞偑丆梡検埶懚揑偱偁傝丆崅楊幰傗懡嵻暪梡偺応崌偵儕僗僋偑崅傑傞丅
SNRI 偺僨儏儘僉僙僠儞丆SSRI 偺僷儘僉僙僠儞丆僼儖儃僉僒儈儞側偳偼丆搳梌奐巒帪偵埆怱丆怘梸晄怳偺敪尰昿搙偑崅偔丆偦偺懠偺暃嶌梡偲偟偰摢捝丆晄柊丆嫽暠側偳偑偁傞丅僷儘僉僙僠儞偺CYP2D6 慾奞嶌梡偵傛傞憡屳嶌梡偵傕拲堄偑昁梫偱偁傞丅
❷ 峈偗偄傟傫栻
乵嶌梡婡彉丒摿挜乶丂庡側嶌梡婡彉偲偟偰丆
- 恄宱嵶朎枌偺Na亄僠儍僱儖偵嶌梡偟丆Na亄僠儍僱儖傪慾奞偡傞偙偲偵傛傝丆恄宱偺嫽暠傪梷惂偡傞丅
- GABA 庴梕懱偵嶌梡偟丆夁忚側恄宱嫽暠傪梷惂偡傞丅
- 嫽暠惈恄宱偺慜僔僫僾僗偵懚嵼偡傞揹埵埶懚惈Ca2亄僠儍僱儖偺兛2兟僒僽儐僯僢僩偵寢崌偟丆Ca2亄棳擖傪梷惂偟丆恄宱嫽暠傪梷偊傞丅
側偳偑峫偊傜傟傞丅
偝傜偵丆儀儞僝僕傾僺儞宯偱峈偗偄傟傫栻偲偟偰傕巊梡偝傟傞僋儘僫僛僷儉偼丆GABA 僯儏乕儘儞偺嶌梡傪摿堎揑偵憹嫮偡傞丅
峈偗偄傟傫栻偼丆栻暔憡屳嶌梡傪棃偡栻嵻偑懡偔丆懡嵻暪梡偵拲堄傪梫偡傞丅僾儗僈僶儕儞丆僈僶儁儞僠儞偼娞憻偱偺戙幱傪傎偲傫偳庴偗側偄偨傔丆栻暔憡屳嶌梡偺塭嬁傪庴偗偵偔偄偲偄偆棙揰偑偁傞丅
乵暃嶌梡乶丂峈偗偄傟傫栻偵嫟捠偡傞暃嶌梡偲偟偰柊婥丆傆傜偮偒偑偁傞偑丆暃嶌梡偺敪尰傪梷偊傞偨傔偵偼掅梡検偐傜奐巒偡傞偙偲偑朷傑偟偄丅摿挜揑側暃嶌梡偲偟偰偼丆埲壓偺傕偺偑偁傞丅
僶儖僾儘巁偱偼娞婡擻忈奞丆崅傾儞儌僯傾寣徢傪棃偡偙偲偑偁傞偨傔丆掕婜揑側娞婡擻専嵏傪峴偄丆堄幆忈奞傪擣傔偨応崌偵偼寣拞傾儞儌僯傾抣偺應掕傪峴偆丅
僇儖僶儅僛僺儞偱偼丆怱巋寖揱摫偺梷惂嶌梡偑偁傞偨傔丆廳撃側怱忈奞乮戞Ⅱ搙埲忋偺朳幒僽儘僢僋丆崅搙偺彊柆乯偺偁傞姵幰偼嬛婖偱偁傞傎偐丆崪悜梷惂偑擣傔傜傟傞偨傔壔妛椕朄丒曻幩慄帯椕丒慡恎惈崪揮堏偱斈寣媴尭彮傪棃偟偰偄傞姵幰偱偼尨懃偲偟偰巊梡偟側偄丅
僾儗僈僶儕儞丆僈僶儁儞僠儞偱偼丆柊婥丆傔傑偄側偳偑偁傝丆恡婡擻掅壓偵傛傝攔煏偑抶墑偝傟傞偨傔丆恡婡擻偵傛傝搳梌検偺挷愡偑昁梫偱偁傞丅
❸ 嬊強杻悓栻丒峈晄惍柆栻
乵嶌梡婡彉丒摿挜乶丂儕僪僇僀儞丆儊僉僔儗僠儞偼丆Vaughan-Williams 峈晄惍柆栻偺僋儔僗嘥b 孮偵埵抲偯偗傜傟偰偍傝丆Na亄僠儍僱儖傪幷抐偡傞偲偄偆揹婥惗棟妛揑側嶌梡婡彉偑峫偊傜傟偰偄傞丅枛徑恄宱偺恄宱忈奞惈醬捝偱偼丆懝彎偟偨恄宱偵偍偄偰Na亄僠儍僱儖偺検丆幙偑曄壔偟丆惓忢偱偼側偄Na亄僠儍僱儖偑敪尰偟恄宱偑夁晀偵側傞偙偲偑娭學偟偰偄傞丅慡恎搳梌偝傟偨儕僪僇僀儞偼丆惓忢側恄宱揱払傪幷抐偣偢偵丆偙傟傜偺Na亄僠儍僱儖傪幷抐偟丆恄宱偺夁晀斀墳傪梷惂偡傞丅傑偨丆C 慄堐偐傜偺巋寖偵傛傝妶惈壔偡傞愐悜屻妏偺僯儏乕儘儞偺妶摦惈傪梷偊丆愐悜屻崻恄宱愡偺敪壩傪梷偊傞偙偲偵傛傝丆夁忚側妶摦揹埵傪梷惂偡傞丅
儊僉僔儗僠儞偼丆娞弶夞捠夁岠壥偑彫偝偔丆挵娗偐傜偺媧廂偑椙岲偱偁傝丆惗懱撪棙梡棪偑栺90亾偲崅偄偨傔偵丆宱岥偱岠壥偑婜懸偱偒傞丅
乵暃嶌梡乶丂儕僪僇僀儞偼丆巋寖揱摫梷惂嶌梡偲怱嬝梷惂嶌梡傪桳偡傞偨傔丆廳撃側巋寖揱摫忈奞偺偁傞姵幰偵偼嬛婖偱偁傞丅儕僪僇僀儞偺怱寣娗宯偺暃嶌梡偲偟偰偼丆寣埑掅壓丆彊柆側偳偑偁傞丅廳戝側暃嶌梡偲偟偰偼丆拞悤恄宱宯偺徢忬乮晄埨丆嫽暠丆帹柭丆怳愴丆枛徑抦妎堎忢側偳乯偑偁傝丆崅擹搙偱偼堄幆徚幐丆慡恎偗偄傟傫傪堷偒婲偙偡偙偲傕偁傞丅峈晄惍柆栻偲偟偰偺桳岠堟偼丆1.5乣5.0兪g/mL 偲偝傟丆10兪g/mL 埲忋偱暃嶌梡偑敪尰偟傗偡偔側傞丅偙傟傜偺暃嶌梡偼梡検埶懚揑偱偁傞偑丆慡恎忬懺偺掅壓偟偨偑傫姵幰偱偼彮検偱傕惗偠傞偙偲偑偁傞偺偱丆廫暘側娤嶡傪峴偆丅傑偨杮嵻偼CYP3A4 偱戙幱偝傟丆妶惈傪桳偡傞戙幱暔偺拁愊偑恄宱撆惈傪堷偒婲偙偡丅
儊僉僔儗僠儞傕傑偨丆廳撃側巋寖揱摫忈奞偺偁傞姵幰偵偼嬛婖偱偁傞丅偦偺懠偺暃嶌梡偲偟偰偼丆埆怱丒歲揻丆怘梸晄怳丆堓晹晄夣徢忬側偳偺徚壔婍徢忬偺弌尰昿搙偑崅偄丅
❹ NMDA 庴梕懱漢峈栻
乵嶌梡婡彉丒摿挜乶丂NMDA乮N-methyl-D-aspartate乯庴梕懱偼丆僌儖僞儈儞巁庴梕懱偺僒僽僞僀僾偺堦偮偱丆拞悤惈姶嶌仏1 傗儚僀儞僪傾僢僾尰徾仏2 偺宍惉側偳丆捝傒側偳偺怤奞忣曬揱払偵廳梫側栶妱傪壥偨偟偰偄傞乮Ⅱ-1-1-3 恄宱忈奞惈醬捝偺崁嶲徠乯丅恄宱忈奞惈醬捝偺敪惗偵偼丆嫽暠惈恄宱揱払暔幙偱偁傞僌儖僞儈儞巁偑梀棧偝傟丆NMDA 庴梕懱傪妶惈壔偡傞偙偲傕娭梌偟偰偄傞丅僆僺僆僀僪偺捔捝懴惈仏3 偵漢峈偟丆捔捝岠壥傪憹嫮偡傞丅
働僞儈儞偼丆廬棃丆杻悓栻偲偟偰巊梡偝傟偰偒偨偑丆懷忬醰怾屻恄宱捝丆尪巿捝傪娷傓偝傑偞傑側恄宱忈奞惈醬捝傪娚榓偡傞丅杮朚偱擖庤壜擻側働僞儈儞惢嵻偼丆惷拲丒嬝拲惢嵻偱偁傝丆2007 擭偐傜杻栻巜掕偲側偭偨丅
偦偺懠丆捔奝栻偺僨僉僗僩儘儊僩儖僼傽儞丆峈僷乕僉儞僜儞栻丒峈A 宆僀儞僼儖僄儞僓僂傿儖僗栻偺傾儅儞僞僕儞丆擼弞娐丒戙幱夵慞栻偱偁傞僀僼僃儞僾儘僕儖側偳偑偙偺暘椶偵娷傑傟傞偑丆椪彴忋偺桳梡惈偵偮偄偰偺抦尒偼尷傜傟偰偄傞丅
乵暃嶌梡乶丂働僞儈儞偼丆擼埑傪槾恑偝偣傞偨傔丆擼寣娗忈奞丆崅寣埑丆擼埑槾恑徢丆廳徢偺怱戙彏晄慡偺姵幰偵偼嬛婖偱偁傞丅庡側暃嶌梡偲偟偰丆柊婥丆傆傜偮偒丆傔傑偄偑偁傞丅廳戝側暃嶌梡偲偟偰媫惈恡晄慡丆屇媧梷惂丆偗偄傟傫側偳偑偁傝丆摿挜揑側徢忬偲偟偰丆尪妎丆埆柌側偳偺拞悤惈嶌梡偑抦傜傟傞丅
仏1丗拞悤惈姶嶌
嫽暠忬懺偵偁傞枛徑恄宱偐傜偼擇師僯儏乕儘儞偵巋寖傪揱偊傞嫽暠惈傾儈僲巁偺僌儖僞儈儞巁乮Glu乯偑曻弌偝傟傞偑丆姶嶌偝傟偨枛徑恄宱偐傜偼Glu 偵壛偊偰僒僽僗僞儞僗P 傗僯儏乕儘僉僯儞A 偲偄偭偨僞僉僉僯儞傕曻弌偝傟傞丅偙傟偵傛傝揹埵埶懚惈Ca2亄僠儍僱儖偐傜Ca2亄偑曻弌偝傟傞偲NMDA 庴梕懱偑妶惈壔偡傞丅偦偺寢壥丆恄宱嵶朎偑夁晀壔偟丆捝妎夁晀傗傾儘僨傿僯傾偑敪惗偡傞丅嶲徠丅
仏2丗儚僀儞僪傾僢僾尰徾
孞傝曉偟捝傒偺巋寖偑壛傢傞偲丆捝妎恄宱廔枛乮愐悜屻妏晹乯偱揱払暔幙曻弌偑憹壛偟丆嵟弶偺捝傒忣曬偑師偵憲傜傟偰偔傞捝傒忣曬傪憹暆偟丆師戞偵捝傒偑憹嫮偡傞尰徾丅
仏3丗捔捝懴惈
弶婜偵搳梌偝傟偰偄偨栻暔偺梡検偱摼傜傟偰偄偨捔捝岠壥偑帪娫宱夁偲偲傕偵尭戅偟丆摨偠捔捝岠壥傪摼傞偨傔偵偼傛傝懡偔偺梡検偑昁梫偵側傞偙偲丅
❺ 拞悤惈嬝抩娚栻
乵嶌梡婡彉丒摿挜乶丂僶僋儘僼僃儞偼丆GABAB庴梕懱仏4 偺嶌摦栻偱偁傝丆嶰嵆恄宱捝丆嬝醶弅丆嬝醶惈醬捝側偳偵巊梡偝傟傞丅嶌梡婡彉偲偟偰偼丆僔僫僾僗慜偺僇儖僔僂儉擹搙傪掅壓偝偣丆嫽暠惈傾儈僲巁偺曻弌傪尭彮偝偣丆屻僔僫僾僗偱偼僇儕僂儉偺揱摫惈傪憹壛偝偣偰恄宱偺夁暘嬌傪婲偙偡丅
乵暃嶌梡乶丂僶僋儘僼僃儞偺庡側暃嶌梡偼丆傔傑偄丆柊婥丆徚壔婍徢忬偱偁傞丅拞悤恄宱宯偵嶌梡偡傞偨傔丆廳戝側暃嶌梡偲偟偰丆堄幆忈奞丆屇媧梷惂側偳偑偁傞丅恡攔煏偱偁傞偨傔恡婡擻掅壓帪偵拲堄偑昁梫偱偁傝丆傑偨撍慠偺拞巭偵傛傝丆棧扙徢岓孮乮尪妎丆嫽暠丆偗偄傟傫側偳乯傪掓偡傞偙偲偑偁傞偨傔丆拞巭偵嵺偟偰偼慟尭偑昁梫偱偁傞丅
仏4丗GABAB庴梕懱
拞悤恄宱宯僯儏乕儘儞傗惎忬嵶朎偵敪尰偟偰偄傞兞-傾儈僲棌巁乮GABA乯庴梕懱偺堦偮丅GABAB庴梕懱偼G 抈敀嫟栶宆偲偟偰婡擻偡傞丅GABAB庴梕懱傪夘偟偰嶌梡偡傞栻嵻偵嶰娐宯峈偆偮栻側偳偑偁傞丅
❻ 僐儖僠僐僗僥儘僀僪
乵嶌梡婡彉丒摿挜乶丂崪揮堏捝丆庮釃偵傛傞恄宱埑敆丆娭愡捝丆摢奧撪埑槾恑丆娗峯憻婍偺暵嵡側偳偵傛傞捝傒偵巊梡偝傟傞丅嶌梡婡彉偼柧妋偱偼側偄偑丆捝傒傪姶抦偡傞晹埵偺晜庮偺寉尭丆僐儖僠僐僗僥儘僀僪斀墳惈偺庮釃偺弅彫丆怤奞庴梕婍偺妶摦惈掅壓乮僾儘僗僞僌儔儞僕儞丆儘僀僐僩儕僄儞傪庡偲偡傞墛徢暔幙偺寉尭乯側偳偲偝傟傞丅
捔捝曗彆栻偲偟偰偼丆嶌梡帪娫偑挿偔丆揹夝幙嶌梡仏1 偑斾妑揑庛偄儀僞儊僞僝儞丆僨僉僒儊僞僝儞偑峀偔巊梡偝傟傞丅僾儗僪僯僝儘儞傪戙懼栻偲偟偰巊梡偡傞偙偲傕偁傞丅
乵暃嶌梡乶丂庡側暃嶌梡偲偟偰丆岥峯僇儞僕僟徢丆崅寣摐丆徚壔惈捵釃丆堈姶愼徢丆枮寧條婄杄丆崪慹偟傚偆徢丆惛恄恄宱徢忬乮偣傫栂傗梷偆偮乯側偳偑偁傞丅搳梌偑挿婜偵媦傇偵廬偄丆暃嶌梡偺昿搙傕崅偔側傞偨傔丆崅楊幰傗崌暪徢傪桳偡傞僴僀儕僗僋姵幰偺応崌丆惗柦梊屻傪娷傔偰搳梌奐巒帪婜偵偮偄偰偺廫暘側専摙偑昁梫偱偁傞丅
仏1丗揹夝幙嶌梡
揹夝幙偺僶儔儞僗傪挷惍偡傞嶌梡丅僗僥儘僀僪偼寣拞偺Na 傪憹壛偝偣丆K 傪尭彮偝偣傞嶌梡偑偁傞丅Na 偺憹壛偼寣埑偺忋徃丆K 偺尭彮偼扙椡姶傗怱晄慡側偳傪堷偒婲偙偡偙偲偑偁傞丅嶌梡偺嫮庛偼僗僥儘僀僪偺庬椶偵傛傝堎側傞丅
❼ 儀儞僝僕傾僛僺儞宯峈晄埨栻
乵嶌梡婡彉丒摿挜乶丂儀儞僝僕傾僛僺儞宯峈晄埨栻偺嶌梡婡彉偲偟偰偼丆戝擼曈墢宯丆帇彴丆帇彴壓晹側偳偵嶌梡偟捔惷嶌梡傪傕偨傜偡偲偝傟偰偄傞丅偙偺嵺偵丆摿堎揑側儀儞僝僕傾僛僺儞庴梕懱乮GABAA庴梕懱仏2-Cl-僠儍僱儖暋崌懱乯偵嶌梡偟丆梷惂惈恄宱揱払暔幙偱偁傞GABAA偺恊榓惈傪崅傔丆Cl-僠儍僱儖偺奐岥偵傛傝夁暘嬌傪婲偙偟丆恄宱枌偺嫽暠惈偑梷惂偝傟傞丅傑偨丆愐悜斀幩梷惂偵傛傝丆嬝偺夁嬞挘傪娚榓偡傞偲偝傟偰偄傞丅僕傾僛僷儉偼丆嬝醶弅偺捝傒偵巊梡偝傟傞丅
乵暃嶌梡乶丂僕傾僛僷儉偺庡側暃嶌梡偼丆柊婥丆傆傜偮偒丆嬝抩娚嶌梡偱偁傞丅摿偵崅楊幰偵懳偟偰丆僕傾僛僷儉偺傛偆側挿帪娫嶌梡宆栻傪巊梡偡傞応崌偼丆嶌梡偑慗墑偡傞偙偲偑偁傞偺偱丆彮検偐傜奐巒偟丆廫暘側娤嶡偑昁梫偱偁傞丅
仏2丗GABAA庴梕懱
兞-傾儈僲棌巁乮GABA乯庴梕懱偺堦偮丅GABAA庴梕懱偼Cl-僠儍僱儖宆偲偟偰婡擻偡傞丅儀儞僝僕傾僛僺儞宯栻嵻側偳偼GABAB庴梕懱傪夘偟偰嶌梡偟丆捔惷丆峈偗偄傟傫丆峈晄埨側偳偺嶌梡傪傕偮丅
❽ 價僗儂僗儂僱乕僩丆僨僲僗儅僽側偳偺bone-modifying agents乮BMA乯
乵嶌梡婡彉丒摿挜乶丂崪揮堏捝偵巊梡偝傟傞價僗儂僗儂僱乕僩惢嵻偺婎杮崪奿偼丆柍婡偺僺儘儕儞巁墫偺桿摫懱偱偁傝丆攋崪嵶朎偺妶摦傪梷惂偟丆崪媧廂傪慾奞偡傞偙偲偵傛傝捔捝岠壥傪摼傞丅岠壥偼梡検埶懚惈偱偁傞丅
僨僲僗儅僽偼丆RANKL乮receptor activator of nuclear factor-kappa B ligand乯偲寢崌偟丆攋崪嵶朎偍傛傃偦偺慜嬱嵶朎枌忋偵敪尰偡傞RANK 傊偺RANKL 偺寢崌傪摿堎揑偵慾奞偡傞暘巕昗揑栻乮僸僩宆峈RANKL 儌僲僋儘乕僫儖峈懱乯偱偁傞丅RANKL 宱楬傪夘偟偨攋崪嵶朎偺宍惉丆妶惈丆惗懚傪梷惂偟丆崪攋夡偵婲場偡傞昦揑崪愜側偳偺崪娭楢帠徾乮skeletal related event丟SRE乯偺敪尰傪梷惂偡傞偲偝傟丆捔捝曗彆栻偵暘椶偡傞偐斲偐偼媍榑偺梋抧偑偁傞偑丆崪捝夵慞偵娭梌偡傞偲偄偆堄枴偱曗懌揑偵晅婰偡傞丅
乵暃嶌梡乶丂價僗儂僗儂僱乕僩偺庡側暃嶌梡偼丆埆怱丆傔傑偄丆敪擬丆媫惈恡晄慡側偳偱偁傞偑丆廳撃側暃嶌梡偲偟偰妠崪夡巰丒妠崪崪悜墛偑弌尰偡傞偙偲偑偁傞丅曬崘偝傟偨徢椺偺傎偲傫偳偑敳帟側偳偺帟壢張抲傗嬊強姶愼偵娭楢偟偰敪尰偟偰偍傝丆埆惈庮釃丆壔妛椕朄丆僐儖僠僐僗僥儘僀僪帯椕丆曻幩慄帯椕丆岥峯撪偺晄塹惗丆帟壢張抲偺婛墲楌偑梫場偲偟偰嫇偘傜傟傞丅昁梫偵墳偠偰揔愗側帟壢専嵏傪峴偄丆杮嵻搳梌拞偼丆怤廝揑側帟壢張抲偼偱偒傞尷傝旔偗傞偙偲丆姵幰偵廫暘側愢柧傪峴偄丆堎忢偑擣傔傜傟偨応崌偵偼丆捈偪偵帟壢丒岥峯奜壢傪庴恌偡傞傛偆拲堄偡傞偙偲偑昁梫偱偁傞丅傑偨丆媫懍揰揌偵傛傝恡晄慡偑弌尰偡傞偙偲偑偁傞偨傔丆搳梌懍搙偵傕拲堄偟丆搳梌奐巒慜偵恡婡擻専嵏傪幚巤偟丆恡婡擻偵傛傞搳梌検傪挷愡偡傞丅
僨僲僗儅僽偺妠崪夡巰丒妠崪崪悜墛側偳偼丆價僗儂僗儂僱乕僩惢嵻偲摨條偱偁傞偑丆嵟傕拲堄偡傋偒偼廳撃側掅僇儖僔僂儉寣徢偺弌尰偱偁傞丅巰朣椺偵帄偭偨徢椺偑曬崘偝傟偨偙偲傛傝昿夞偵寣塼専嵏傪峴偄丆寣惔曗惓僇儖僔僂儉抣偑崅抣偱側偄尷傝丆僇儖僔僂儉偍傛傃價僞儈儞D 偺宱岥曗廩偺傕偲偵搳梌偡傞傛偆丆寈崘慬抲偲側偭偰偄傞丅
❾ 偦偺懠
徚壔娗暵嵡偵傛傞捝傒偵懳偡傞僆僋僩儗僆僠僪丆僽僠儖僗僐億儔儈儞廘壔暔側偳丆摿掕偺捝傒偵巊梡偡傞忋婰埲奜偺栻嵻偵偮偄偰偼嘨復悇彠偺杮暥傪丆嶌梡婡彉丒摿挜偵偮偄偰偼懠崁傪嶲徠偝傟偨偄丅
乮媣尨丂岾乯
亂暥丂專亃
1乯 Finnerup NB, Otto M, McQuay HJ, et al. Algorithm for neuropathic pain treatment丗an evidencebased proposal. Pain 2005丟118丗289-305
2乯 Wiffen PJ, Collins S, McQuay HJ, et al. Anticonvulsant drugs for acute and chronic pain. Cochrane Database Syst Rev 2005丟Issue 3
3乯 Saarto T, Wiffen PJ. Antidepressants for neuropathic pain. Cochrane Database Syst Rev 2007丟Issue 4
4乯 Bell RF, Eccleston C, Kalso E. Ketamine as an adjuvant to opioids for cancer pain. Cochrane Database Syst Rev 2003丟Issue 1
5乯 Challapalli V, Tremont-Lukats IW, McNicol ED, et al. Systemic administration of local anesthetic agents to relieve neuropathic pain. Cochrane Database Syst Rev 2005丟Issue 4
6乯 Wong R, Wiffen PJ. Bisphosphonates for the relief of pain secondary to bone metastasis. Cochrane Database Syst Rev 2002丟Issue 2
7乯 Eisenberg E, McNicol E, Carr DB. Opioids for neuropathic pain. Cochrane Database Syst Rev 2006丟Issue 3
8乯 Wiffen PJ, McQuay HJ, Edwards JE, et al. Gabapentin for acute and chronic pain. Cochrane Database Syst Rev 2005丟Isuue 3
亂嶲峫暥專亃
9乯 Lussier D, Portenoy RK. Adjuvant analgesics in pain management. Doyle D, Hanks GWC, Cherny NI, Calman K eds. Oxford Textbook of Palliative Medicine. 3rd ed, Oxford University Press, 2003, p349